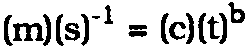MEASURING THE RATE OF ATMOSPHERIC CORROSION IN MICROCLIMATESHARALD BERNDT
2 MEASURING THE CORROSIVENESS OF MICROCLIMATES CREATED BY WOOD AND WOOD PRODUCTSTHE PROBLEMS occurring with objects stored in wood-based containers have been reviewed previously (FitzHugh and Gettens 1971; Berndt 1987). Typical degradation processes, for example, formation of efflorescence on calcareous materials (FitzHugh and Gettens 1971) and lead corrosion (Oddy 1973, 1975; Blackshaw and Daniels 1978; Blackshaw 1982; Padfield 1982; FitzHugh and Gettens 1971; Arni et al. 1965a, 1965b; Packman 1960; Clarke and Longhurst 1961; Packman 1957; Donovan and Stringer 1969, 1971; Carpenter and Hatchfield 1985; Hatchfield and Carpenter 1986; Leveque 1986; Berndt 1987; Thomson 1978), are catalyzed by organic acids. For the conservator, catalytic action is especially undesirable because small amounts of the catalyst may have significant degradative effects and because the deleterious agent is not consumed in the degradation process. The latter is useful for an analysis of the environment by deterioration rate measurements because the exposed test material will change the environment only negligibly. The undesirable action of organic acids in the museum environment stems from the high solubility of most of their metal salts (Weast 1979). While calcium carbonate, the main constituent of sea shells and other calcareous materials, is insoluble in water, calcium acetate is soluble. Dissolution of calcium acetate, relocation, and redeposition on drying caused the calclacite efflorescence seen on several museum objects (FitzHugh and Gettens 1971). Similarly, catalysis of metal corrosion by organic acid vapors proceeds by metal oxide dissolution in the anodic areas of the metal surface. The dissolved organic acid salts migrate to the relatively alkaline cathodic areas, where they hydrolyze and form metal hydroxide (Donovan and Stringer 1971; Scully 1975). The hydroxide may become less soluble by carbonation. In lead corrosion, this leads to the formation of basic lead carbonate, or white lead, as the principal product (Oddy 1973, 1975; FitzHugh and Gettens 1971; Donovan and Stringer 1971; Berndt 1987). Of the organic acid vapor-induced degradation processes mentioned above, only metal corrosion is readily quantifiable. Calclacite efflorescence on calcareous materials involves a relocation of material only, which depends largely on condensation/evaporation cycles and is not sensitive enough to the amount of organic acids present. Zinc and lead are the metals most susceptible to organic acid vapor corrosion (Donovan and Stringer 1969, 1971). Comparing the solubilities of zinc and lead chlorides to the solubilities of the respective organic acid salts (Weast 1979) suggests that zinc is also very vulnerable to hydrochloric acid vapors while lead is not. The specificity of lead to corrosion by organic acids makes it the metal of choice for an atmospheric corrosiveness indicator. Metal corrosion rate is measured as the amount of metal converted to metal salt per unit time and per unit area of exposed surface. To be a useful indicator, corrosion rate has to increase monotonically with increasing organic acid concentration. This is likely unless the corrosion mechanism changes at some concentration. A change in corrosion mechanism usually leads to a change in the corrosion product formed (Clarke and Metal corrosion rate is commonly determined by exposing a cleaned test coupon with known surface area and weight to the conditions of interest for a length of time, removing the salt formed by an appropriate method, and determining the weight loss. Dividing the weight loss by the exposed surface area and the exposure time gives the corrosion rate (ASTM 1981). If the salt formed is the same under all conditions and if it does not separate from the test coupon during the test period, the weight gain on exposure can be used for corrosion rate determination. Following weight gain has the advantage of eliminating the often tedious cleaning step at the end of the exposure period. It also allows simple interpretation of continuous weight measurements. The standard method of calculating corrosion rate, discussed above, implies the assumption that the rate is constant with time, that is, that equal amounts of metal corrode in any time interval of the same length. That is usually not the case. Previous experience with corrosion in the presence of constant concentrations of organic acids showed that the corrosion rate decreased with time (Clarke and Longhurst 1961). As salt forms on the metal surface, reactants for the corrosion reaction have to diffuse through an increasingly thick salt layer to reach the reaction sites. The diffusion rate may become limiting for the corrosion rate. The simplest mathematical model for a decreasing corrosion rate is:
Besides the organic acid concentration in the test atmosphere, lead corrosion rate will depend on the temperature and humidity of the environment. For various reasons, the organic acid vapor concentration in the environment of wood and wood products will also be a function of temperature and humidity. It is desirable to keep the test period short so that organic acid concentration will not change due to secondary processes and so that results will be achieved quickly. The conditions used in this series of tests, 50�C and 80% RH, seemed the best compromise for accelerating the corrosion reaction without changing the mechanisms leading to the organic acid release or the lead corrosion. In atmospheres of 100% RH, small drops in temperature would cause condensation of water inside the wood, which can lead to biodeterioration (Schniewind 1989) and accompanying unpredictable changes in the acid release behavior. At temperatures in excess of 65�C, wood may become subject to thermal degradation processes (Wilcox 1989). For the conditions chosen, exposure times of two weeks were sufficient. More corrosive atmospheres and longer exposure times led to a loss of integrity of the test coupons. |