THE USE OF MINERALOGICAL DATA IN INTERPRETATION OF LONG-TERM MICROBIOLOGICAL CORROSION PROCESSES: SULFIDING REACTIONSM. B. MCNEIL, & B. J. LITTLE
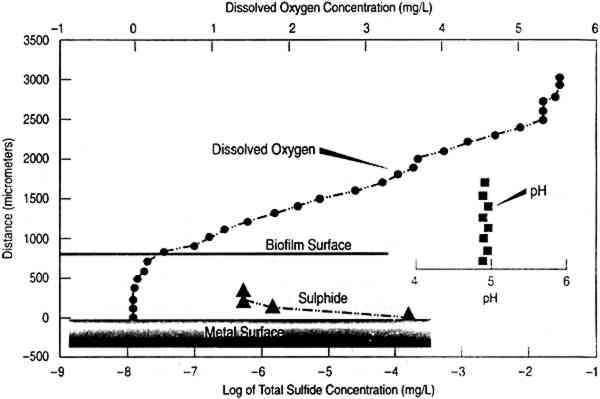
ABSTRACT—Immersed or buried objects are covered with a biofilm. The microbiological consortia in the biofilms play an important role in determining the corrosion or alteration of such objects, particularly archaeological artifacts. Mineral deposits on the surface of these objects can provide useful insights into the processes of degradation and, in cases of severe degradation, sometimes into the nature of the original objects. This article reviews results of mineralogical studies on corroded objects in general, with an emphasis on sulfides. TITRE—L'Utilisation de Donn�es Min�ralogiques dans L'Interpr�tation des Processus de Corrosion Microbiologique � Long Terme: Le Sulfurage. R�SUM�—Les objets immerg�s ou enfouis sous terre sont recouverts d'un film biologique. Le groupement microbiologique dans ce film joue un r�le important dans l'�volution de la corrosion et les autres changements qui se produisent, en particulier sur les objets arch�ologiques. Les d�p�ts min�raux � la surface de ces objets peuvent aider � comprendre le processus de d�gradation et donner des indices sur la nature originale des objets dans les cas de d�gradation tr�s avanc�e. Dans cet article, les r�sultats d'�tudes min�ralogiques sur des objets corrod�s sont pass�s en revue de fa�on g�n�rale, avec une attention particuli�re sur les sulfures. TITULO—La utilizaci�n de datos mineral�gicos en la interpretaci�n de procesos de corrosi�n microbiol�gica a largo plazo: reacciones de sulfuraci�n. RESUMEN—Los objetos sumergidos o enterrados est�n cubiertos por una pel�cula biol�gica. El conjunto microbiol�gico en estas pel�culas juega un importante papel en el proceso de corrosi�n o alteraci�n de tales objetos, particularmente los objetos arqueol�gicos. Los dep�sitos minerales sobre la. superficie de estos objetos puede dar luces sobre el proceso de degradaci�n, y en los casos de degradaci�n severa, nos da informaci�n sobre la naturaleza original de los objetos. Este articulo hace un an�lisis de los datos de estudios mineral�gicos en objetos con corrosiones diversas, y en especial con corrosiones de sulfuraci�n. 1 INTRODUCTIONObjects exposed to corrosive environments for long periods of time, whether they are meteorites, archaeological objects, infrastructure components, or minerals exposed to weathering, frequently show degradation of a nature that cannot be duplicated in laboratory experiments, either in degree or sometimes even in kind. Some laboratory experiments have carefully exposed characterized specimens to known environments for decades (Romanoff 1957); however, for practical purposes the laboratory time frame is five years or less. In this article, “long-term” will be applied to time frames sufficiently long that the areas where reactions take place may be regarded as macroscopic regions with local equilibrium, with the overall development of individual reactions controlled by mass transport considerations rather than local kinetics. Central to this reasoning is the applicability of Pourbaix diagrams (Garrels and Christ 1965; Pourbaix 1966) to predict not only regions of immunity and passivity but also the identities of the species produced by corrosion. The use of diagrams is complemented by Ostwald's principle (Ostwald 1906) that when a component (e.g., a metal) reacts with an element in its environment (e.g., reduced sulfide species), the normal succession of reaction products is initially Sulfiding corrosion is a common consequence of biofouling, because sulfur compounds are common in the biosphere and ubiquitous in the industrialized world. Sulfate-reducing bacteria (SRB), a diverse group of anaerobic bacteria isolated from a variety of sulfur-containing environments (Postgate 1979; Pfennig et al. 1981), are capable of reducing sulfate and similar oxidized sulfur species to sulfides. SRB are routinely isolated from seawater where the concentration of sulfate is typically 25 mM (Postgate 1979). Even though seawater is generally aerobic (typical oxygen concentrations above the thermocline, the temperature change generally regarded as dividing near-surface from deep seawater, are in the range of 4–6 ppm, equivalent to .1–.15 mM), anaerobic micro-organisms survive in anaerobic microniches until conditions are suitable for their growth (Staffeldt and Kohler 1973; Costerton and Geesey 1986). If the aerobic respiration rate within a biofilm is greater than the oxygen diffusion rate, the metal-biofilm interface can become anaerobic and provide a niche for sulfide production by SRB (Little et al. 1990). Conversion of metals to sulfide minerals by SRB has been studied since the late 1800s (Daubree 1862; de Gouvernain 1875). Baas-Becking and Moore (1961) list sulfides produced by Desulfovibrio desulfuricans over a large pH range. In the following sections, SRB sulfide production on pure metal surfaces will be reviewed. The metal interface under the biofilm and corrosion layers will be referred to as base metal to differentiate it from layers of minerals and metal ions that have been derivatized by corrosion reactions. Mineralogical data, thermodynamic stability diagrams (Pourbaix 1966; Wagman et al. 1982), and the simplexity principle for precipitation reactions (Goldschmidt 2 MINERALOGICAL OBSERVATIONS2.1 IRONThe corrosion rate of iron in the presence of hydrogen sulfide is accelerated by formation of iron sulfide minerals (Wikjord et al. 1980) that stimulate the cathodic reaction. Once electrical contact is established, mild steel behaves as an anode and electron transfer occurs through the iron sulfide. In the absence of oxygen, the metabolic activity of SRB causes accumulation of hydrogen sulfide near metal surfaces. This accumulation is particularly evident when metal surfaces are covered with biofilms. Figure 1 shows concentration profiles of sulfide, oxygen, and pH in a biofilm accumulated on the surface of a mild steel corrosion coupon. The concentration of sulfide is highest near the metal surface where iron sulfide forms quickly and covers the steel surface if both ferrous and sulfide ions are available. At low ferrous ion concentrations, adherent and temporarily protective films of iron sulfides are formed on the steel surface, with a consequent reduction in corrosion rate.
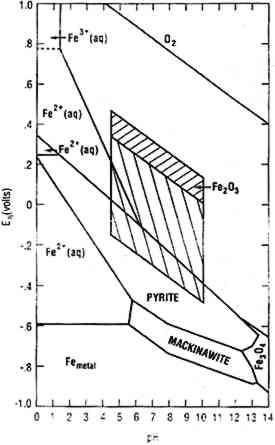
Figure 2 is a stability diagram for an iron-water-reduced sulfur system with lines for 10−6 M ferrous iron and 10−2 M sulfide. For clarity, pyrite (FeS2) and mackinawite (Fe9S8) are the only sulfides indicated. Mackinawite is a tetragonal mineral that may be unstable altogether but is definitely unstable above 150�C. It cannot be produced by conventional techniques. Cubic iron sulfide (FeS) can be produced artificially. By applying hydrogen sulfide (H2S) pressures in the range of one atmosphere, Berner (1969) produced “tetragonal FeS,” with the same symmetry as mackinawite but containing somewhat less sulfur and with slight but systematic differences in lattice parameter. Presumably, further increases in H2S pressure could produce material equivalent to natural mackinawite. Parallelograms superimposed on the diagram are bounded by the highest and lowest pH values commonly found in fresh and saline surface waters. The upper portion of the hatched area applies to waters less than 10 m from the surface; the lower (oppositely hatched) area to deeper waters (Garrels and Christ 1965). The region of stability of mackinawite can be seen to be wholly outside the region defined by surface water conditions, excluding waters influenced by peat bogs, coal mines, volcanic activity, and industrial effluents. Because of its metastability, mackinawite is frequently observed on objects corroded for relatively brief periods, and less frequently on archaeological artifacts.
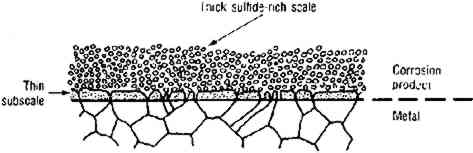
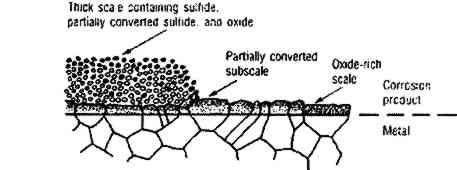
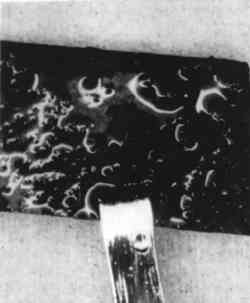
During corrosion of iron and steel in the presence of SRB, a thin (approximately 1 μm), adherent layer of “tarnish” is first formed. This was originally termed “kansite” but has since been identified as mackinawite. As it thickens, the layer becomes less adherent. If ferrous ion concentration in the electrolyte is low, mackinawite alters to greigite, a thiospinel with the formula Fe3S4. (For a more detailed discussion of the older work on these processes, see Trudinger and Swaine 1979, 342–56). If ferrous ion concentration is high, mackinawite is accompanied by green rust 2, a complex In summary, mackinawite is easily produced from iron and iron oxides by consortia of micro-organisms that include SRB. The presence of mackinawite in corrosion products formed in shallow water environments with the exclusions previously delineated is proof that the corrosion was SRB-induced. Recent work indicates that on continued exposure to SRB mackinawite alters to smythite (Fe9S11), to greigite (Fe3S4), to pyrrhotite (FeS1+x), and finally to pyrite (FeS2) (McNeil and Little 1990). SRB in thin biofilms on pottery surfaces (Duncan and Ganiaris 1987; Heimann 1989) and silver (McNeil and Mohr 1993) can produce pyrite films from iron-rich waters. SRB can produce pyrite from mackinawite in contact with elemental sulfur (Berner 1969), and thermodynamic analyses indicate that elemental sulfur can coexist with various iron sulfides. The iron sulfides are important archaeologically for another reason: when iron objects that have been subjected to sulfiding corrosion are retrieved from the sea, reactions can occur that are detrimental to the object and especially to any wood left in association with it (MacLeod and Kenna 1990). 2.2 COPPERCuprite (Cu2O), the first product of copper corrosion, forms epitaxially as a direct reaction product of copper with dissolved oxygen or with water molecules (North and Pryor 1970). Cuprite has a high electrical conductivity and permits transport of copper ions through the oxide layer so they can dissolve in the water and reprecipitate. If the water chemistry approximates that of seawater, copper ions reprecipitate as copper hydroxychloride (Cu2(OH)3Cl) (Pollard et al. 1989). Cu2(OH)3Cl has four different crystal structures, one of which, paratacamite, may not be stable at standard temperature and pressure if copper is the only cation (Jambor et al. 1996); matters are further complicated because in corrosion reactions some other polytypes of this compound (atacamite and clinoatacamite) may form pseudomorphically from the metastable form botallackite (Kato and Pickering 1984). The impact of sulfides on the corrosion of copper alloys has received considerable attention, including published reports documenting localized corrosion of copper alloys by SRB in estuarine environments (Little et al. 1988, 1989) and a report of the failure of copper alloys in polluted seawater containing waterborne sulfides that stimulate pitting and stress corrosion cracking (Rowlands 1965). Copper alloys suffer accelerated corrosion attack in seawater containing 0.01 ppm sulfide after a one-day exposure (Gudas and Hack 1979). A porous layer of cuprous sulfide with the general stoichiometry Cu2−xS, 0<x<1 forms in the presence of sulfide ions (Syrett 1980). Copper ions migrate through the layer, react with more sulfide, and produce a thick, black scale (fig. 3a), which can be altered by oxygen from the environment to a complex sulfide-oxide scale (fig. 3b). The sulfide scale does not confer much protection against further attack, but the sulfide-oxide scale provides even less.
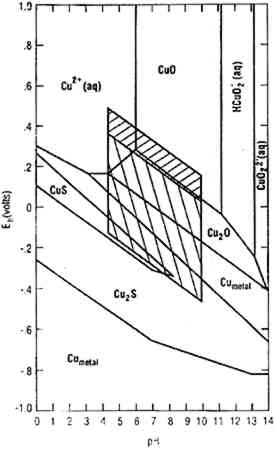
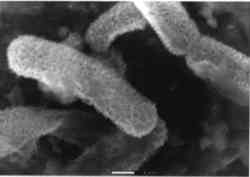
Figure 4 is a stability diagram for copper and its minerals drawn for 10−6 M total dissolved copper and 10−2 M total sulfide. Parallelograms superimposed on the diagram are similar to those described for figure 2 and are appropriate for the analysis of corrosion mineralogy under nonhydrothermal conditions. McNeil et al. (1991) used figure 4 to interpret results from laboratory experiments. They exposed mixed cultures known to contain SRB to copper and copper-nickel alloys in a variety of natural and synthetic waters containing sulfates for 150 days. The pH values of the waters, measured after two weeks, were between 5.5 and 6.8. All copper-containing metals exposed to SRB in isolated cultures and in the natural augmented waters were covered with black, sulfur-rich deposits (fig. 5). The thickness and tenacity of the surface deposits varied among the metals and cultrues. Corrosion products on commercially pure copper were consistently nonadherent. Corrosion products on copper alloys were more adherent and in some cases difficult to scrape from the surface. In all cases, bacteria were closely associated with sulfur-rich deposits, as evident in the scanning electron microscope (SEM) micrographs in figures 6a and 6b; the bacteria are encrusted with copper sulfides. Most SEM micrographs of SRB on copper surfaces indicate a monolayer of cells overlaying a sulfide layer. Transmission electron microscopy has been used to demonstrate that bacteria are intimately associated with sulfide minerals and that on copper-containing surfaces the bacteria were found between alternate layers of corrosion products and attached to base metal (Blunn 1986).
Biomineralogy of copper sulfides has been studied for more than a century (Daubree 1862, de Gouvernain 1875; Baas-Becking and Moore 1961; Mor and Beccaria 1975; Syrett 1977, 1980, 1981; McNeil and Little 1992). The complexity of the resulting observations reflects the complexity of the copper-sulfur system, especially in the presence of alloying elements or iron in the environment (Kostov and Minceva-Stefanova 1981). Both high- and low-temperature polytypes of chalcocite (Cu2S), digenite (Cu9S5), djurleite (Cu1.93S–Cu1.97S), anilite (Cu7S4), spionkopite (Cu39S28), geerite (Cu8S5), and covellite (CuS, generally blue-remaining) have been reported. In long-term corrosion where waters contain significant iron, chalcopyrite (CuFeS2) is a common product Detailed kinetics of individual reactions are not fully understood, and the consequences for corrosion depend on many factors, including mineral morphology and variations of redox and pH with time (McNeil and Mohr 1993). Discussions of alteration kinetics are contained in a number of articles (Baas-Becking and Moore 1961; Roseboom 1966; Craig and Scott 1976; Putnis 1977; Evans 1979). The general phenomenology can be understood by the following approach. Microbial consortia that include SRB produce anoxic, sulfide-rich environments in which the conversion of copper to copper sulfides is thermodynamically favored at a concentration of 10−2 M sulfide (see fig. 4). Reactions appear to proceed as suggested by Ostwald's principle: the first sulfur-poor compounds are converted to sulfur-rich compounds. One would expect a layering effect with covellite on the outside and chalcocite next to unreacted copper metal. This effect has not been studied, and indeed would be very difficult to study on pure copper because of the porous and mechanically unstable corrosion products. In short-term experiments with excess of copper over available sulfur, chalcocite with little or no covellite is formed (McNeil et al. 1991). Covellite is produced if excess sulfide is available, either deliberately provided (Baas-Becking and Moore 1961) or naturally available (Daubree 1862; Mor and Beccaria 1975). The presence of dissolved iron leads to other complications. Not only has chalcopyrite been observed, but also digenite (Baas-Becking and Moore 1961; Mor and Beccaria 1975; North and MacLeod 1986; McNeil et al. 1991), djurleite (Macdonald et al. 1979; McNeil et al. 1991), and the hexagonal high-temperature polytype of chalcocite (McNeil et al. 1991). These observations can be interpreted in terms of the simplexity principle (Goldschmidt 1953): impurities tend to stabilize high-entropy, high-temperature polytypes. Digenite stability is promoted by iron (Craig and Scott 1976) and apparently djurleite by nickel (McNeil et al. 1991). It was observed that corrosion products containing djurleite showed substantial adherence and mechanical stability, while the corrosion products on pure copper, composed of chalcocite with only traces of other minerals, were powdery and nonadherent. It has been argued that if the copper sulfide layer were djurleite, the sulfide layer would be protective (McNeil et al. 1991). Even if such a sulfide film were technically passivating, the mechanical stability of the film is so poor that sulfide films in general cannot be regarded as offering the resistance to further attack associated with good patination. In the There is one class of conditions under which biofilms appear not to produce sulfide minerals. If conditions at the surface of a copper alloy permit precipitation of nantokite (CuCl) under the cuprite layer, the alloy becomes vulnerable to bronze disease (Scott 1990) or pitting corrosion (Lucey 1967), depending on mass transport conditions. The presence of alloying elements does not protect against the formation of nantokite and the consequent bronze disease corrosion (Mond and Cuboni 1893), though it probably affects the kinetics (North and Pryor 1970; Hack et al. 1986). The role of biofilms in the forms of corrosion associated with nantokite is not well understood, though biofilms are frequently observed and acid-producing bacteria could help produce the requisite conditions by increasing acidity in anodic regions and restricting cation transport away from the surface, maintaining higher copper ion concentrations (McNeil and Mohr 1992). Bronze disease and sulfiding microbiologically influenced corrosion are not observed in the same region, perhaps because the local redox environment required to produce bronze disease is too oxidizing for sulfiding. Another possibility is that the copper ion concentrations associated with nantokite, malachite, and the various polytypes of Cu2(OH)3Cl may make the local environment toxic for SRB. The solubilities, while low compared to many copper compounds, are many orders of magnitude higher than those typical of copper sulfides (Brooks et al. 1968; Rose 1969; Livingston 1991). For example, in neutral water with 10−6 M Cl− the concentration of Cu+ in equilibrium with a typical Cu2(OH)3Cl polytype is near 10−3 M, while the concentration of Cu+ in equilibrium with covellite, CuS, is 10−23 M. Metal sulfide solubilities are generally higher than those derived by simple application of solubility products because of the formation of various sulfide complexes. Nonetheless, the equilibrium concentrations of copper ions in contact with sulfide minerals are still orders of magnitude lower than those observed when only oxide minerals are present (Crerar and Barnes 1976; Davies-Colley et al. 1985). 2.3 SILVER
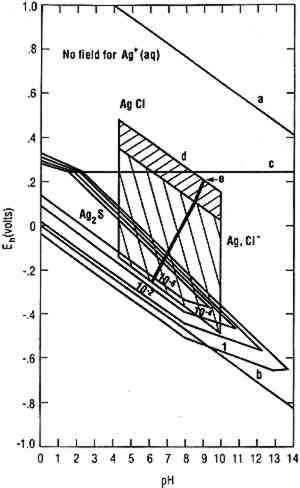
Figure 7 is a silver-water-chloride-sulfur system stability diagram. The different stability regions are annotated with differing sulfide concentrations. Conventionally such diagrams are drawn for a metal ion concentration of 10−6 M. However, in this case there is no region where such a concentration of Ag+ is stable. The limit imposed on Ag+ concentration by the low solubility of Ag sulfide and chloride minerals is
Silver and its alloys are subject to corrosion by reduced sulfur species including H2S, generally of microbiological origin. In air (e.g., in a museum) H2S can be the consequence of biodegradation of sulfur-containing polymeric materials, producing monoclinic acanthite (Ag2S) (Banister 1952; Bauer 1988). Figure 7 indicates the possible thermodynamically stable phases for silver equilibrated with varying total sulfur compositions in 0.46 M sodium chloride (NaCl) (typical for seawater). It is assumed that reduced sulfur species (S2−, HS−, H2S) are in equilibrium. A straightforward type of silver corrosion is conversion of silver to chlorargyrite (AgCl), as indicated above line (c). Below line (c) metallic silver is stable, except for the wedge-shaped areas pointing down and to the right, indicating regions of stability for monoclinic acanthite (Ag2S). The region between the diagonal lines bounding the upper hatched region approximates the effective oxidizing behavior of near-surface, fully aerated seawater (Garrels and Christ 1965). Most shallow sea chemistries fall into this region. Conditions in shallow land burials where the major source of groundwater is rain or surface water percolating through soils are near this region. Chlorargyrite is stable in seawater and chloride-rich shallow-land burial conditions. There are three polymorphs of Ag2S. Monoclinic acanthite is stable up to 176�C (Kracek 1946). Body-centered cubic argentite (Kracek 1946) is stable from 176�C to a temperature between 586�C and 622�C, above which the stable form is a face-centered cubic polymorph (Djurle 1958; Barton 1980). The high-temperature polymorph has never been observed in corrosion. Reactions between Ag2S polymorphs are very fast. Pure body-centered cubic argentite (Ag2S) is not found in nature at standard temperature and pressure, and artificial argentite made with pure silver cannot be quenched to room temperature (Roy et al. 1959). Laboratory data on sulfide attack on silver can be summarized as follows: (1) corrosion of silver by reduced sulfides, whether H2S (Sinclair 1982; Volpe and Peterson 1989) or organic sulfides (Sinclair 1982) produces acanthite; (2) carbon disulfide (CS2) does not produce corrosion; (3) the corrosivity of organic sulfides appears to be controlled by transport mechanisms and thus by vapor pressures; and (4) the rate of sulfidation is strongly affected by ammonia (NH3) and iron dissolved in the silver (Biestek and Drys 1987). Abiotic aqueous corrosion of silver in the presence of reduced sulfur species produces acanthite in bulk (Birss and Wright 1981; Campbell et al. 1982). Argentite is observed when objects made of impure silver (e.g., coins) are corroded in sediments over archaeological periods (Gettens 1963; North and MacLeod 1986). If chloride (Cl−) is present, acanthite or argentite combined with chlorargyrite is formed. These observations support the hypothesis that formation of argentite is limited to precipitation of a silver-copper sulfide by reduced sulfide species. This theory is consistent with the observation that argentite corrosion products are sometimes accompanied by jalpaite (Ag1.5Cu.5S) (North and MacLeod 1986). Argentite made of pure silver is unstable at room temperature; yet there are two reasons why argentite should precipitate during microbiologically influenced corrosion of archaeological objects. Argentite is usually associated with jewelry and coinage containing several percent copper. Argentite, unlike acanthite, can accommodate almost 30% copper in its lattice (Shcherbina 1978). The phenomenon is parallel to the production of akageneite rather than goethite in the corrosion of meteorites, which is attributed to the ability of the akageneite lattice to accommodate significant Cl−, whereas the goethite lattice can accommodate little or none (Buchwald 1977). Precipitation of a mineral from an impure environment favors a loose crystal structure capable of accommodating impurity atoms (Goldschmidt 1953). Argentite formation occurs when an object made of silver-copper alloy is in a water-saturated deposit containing SRB in a biofilm capable of maintaining reducing conditions and bacteria (perhaps ammonia producers) capable of solubilizing silver and copper atoms. A layer of sand or soil restricts the ability of the metal ions to escape, so that concentrations of copper and silver ions within the biofilm rise to levels that cause precipitation. The precipitation of argentite is favored for the reasons given above. Jalpaite forms in regions where the copper concentration is high. Argentite and jalpaite could, in principle, be stabilized by corrosion of pure silver in a copper-rich environment (e.g., silver coins with copper coins), but for practical purposes, the presence of either argentite or jalpaite implies that the silver artifact originally contained significant copper. 2.4 OTHER METALSSRB-induced corrosion of zinc produces a zinc sulfide reported to be sphalerite (ZnS) (Baas-Becking and Moore 1961). SRB on lead carbonates produces galena (PbS) (McNeil and Little 1990). Galena has been found more recently as a lead corrosion product in SRB-induced corrosion of lead-tin alloys (McNeil and Mohr 1993). 3 CONCLUSIONSMicro-organisms within biofilms are capable of maintaining environments at biofilm/metal interfaces that are radically different from the bulk in terms of pH, dissolved oxygen, and concentrations of organic and inorganic species. As a consequence, micro-organisms within biofilms produce minerals and mineral replacement reactions not predicted by thermodynamic arguments based on the chemistry of the bulk medium. Pourbaix diagrams, which correlate corrosion mineralogy with local redox and acidity conditions, can be used to demonstrate that certain minerals (in fact most sulfide minerals) form, in natural waters away from hydrothermal activity, only as a result of microbiological action. There remains the question of how thermodynamic stability diagrams can be applied to answer more detailed questions about archaeological objects. What was the original The issue of using stability diagrams to determine whether corrosion product layers are the result of processes over archaeological times or whether they could be the product of relatively short-term processes has never been comprehensively addressed. One can use stability diagrams and the assumption of local equilibrium to construct trajectories on a stability diagram consistent with mineralogical observations (McNeil and Mohr 1993). One could in principle determine the time scale along the trajectory (Lichtner and Seth 1996), but so far only preliminary work has been done (Walton and McNeil 1994) on bronze disease. Another possible approach is to consider sulfur isotopic analysis. It is plausible that long-term microbiological sulfiding of buried artifacts would produce a sulfur isotope distribution different from that produced by short-term sulfiding corrosion, microbiological or otherwise, because the rate would characteristically be controlled by sulfur availability rather than sulfur conversion. In studying corrosion of materials where corrosion processes can produce a variety of different mineral products, projections of four-dimensional Eh-pH-log anion-log cation diagrams taken normal to the Eh-pH plane (so each point in the diagram corresponds to a Pourbaix diagram) are useful. These are termed modified log activity diagrams (McNeil and Mohr 1992) and are particularly useful in systems where the metal ion has more than one valence state. No effort has been made to use them to interpret sulfiding corrosion. Combination of more extensive thermodynamic-diagram analysis with modeling of electrochemical kinetics (King et al. 1992; Walton and McNeil 1994) mechanisms will permit estimation of rates. This is a critical issue for the appliction of these scientific results to the broader world of archaeological corrosion, and it needs attention. ACKNOWLEDGEMENTSThe authors wish to thank Dr. Lyndsie Selwyn of the Canadian Conservation Institute for careful review of this paper and for numerous valuable suggestions. This work was funded under Program Element 0601153N, Naval Research Laboratory (NRL). NRL Contribution No. NRL/JA/7333-98-0007. REFERENCESBaas-Becking, G. M., and D.Moore. 1961. Biogenic sulfides. Economic Geology56:259–72. Banister, F. A.1952. An unusual synthesis of acanthite crystals. Paper presented at the meeting of the Mineralogical Society of London. Documented in the Ford-Fleischer files of the U. S. Geological Survey, Reston, Va. Barton, M. D.1980. The Ag-Au-S system. Economic Geology75:303–16. Bauer, R.1988. Sulfide corrosion of silver contacts during satellite storage. U. S. Air Force Report SD-TR-88-53/AD-1196 217. Berner, R. A.1969. The synthesis of framboidal pyrite. Economic Geology64:383–84. Biestek, T., and M.Drys. 1987. Corrosion products forming on silver in various corrosive environments. Powlocki Ochronne (Warsaw) 9:2–5. Birss, V. I., and G. A.Wright. 1981. The potentiodynamic formation and reduction of a silver sulfide monolayer on a silver electrode in aqueous sulfide solutions. Electrochimica Acta27:1–7. Blunn, G.1986. Biological fouling of copper and copper alloys. Biodeterioration6:567–75.
Brooks, R. R., B. J.Presley, and I. R.Kaplan. 1968. Trace elements in the interstitial waters of marine sediments. Geochimica et Cosmochimica Acta32:397–414. Buchwald, V. F.1977. The mineralogy of iron meteorites. Philosophical Transactions of the Royal Society (London). A286:453–91. Campbell, G. D., F. J.Lincoln, G. P.Power, and I. M.Ritchie. 1982. The anodic oxidation of silver in sulfide solutions. Australian Journal of Chemistry35:1079–85. Costerton, J. W., and G. G.Geesey. 1986. The microbial ecology of surface colonization and of consequent corrosion. In Biologically induced corrosion, ed.S. C.Dexter. Houston, Tex.: National Association of Corrosion Engineers International. 223–32. Craig, J. R., and S. D.Scott. 1976. Sulfide phase equilibria. In Sulfide mineralogy, ed.P. H.Ribbe. Washington, D. C.: Mineralogical Society of America. CS58–CS75. Crerar, D. A., and H. L.Barnes. 1976. Ore solution chemistry, part 5. Economic Geology71:772–94. Cuthbert, M.1962. Formation of bornite at atmospheric temperature and pressure. Economic Geology57:38–41. Daubree, G. A.1862. Contemporary formation of copper pyrite by the action of hot springs at Bagnes-de-Bigorre. Bulletin de la Societe Geologique de France19:529–32. Davies-Colley, R. J., P. O.Nelson, and K. J.Williamson. 1985. Sulfide control of cadmium and copper concentrations in anaerobic esturine sediments. Marine Chemistry16:173–86. deGouvernain, M.1875. Sulfiding of copper and iron by a prolonge stay in the thermal spring at Bourbon l'Archambault. Comptes Rendus80:1297–1300. Djurle, S.1958. An x-ray study of the system Ag-Cu-S. Acta Chem. Scandinavica12:1427–36. Daucan, S. J., and H.Ganiaris. 1987. Some sulphide corrosion products on copper alloys and lead alloys from London waterfront sites. In Recent advances in the conservation and analysis of artifacts, ed.E. J.Black. London: Summer Schools Press, University of London. 109–18. Evans, H. T.1979. The crystal structures of low chalcocite and djurleite. Zeitschrift f�r Kristallographie150:299–320. Garrels, R. M., and J. C.Christ. 1965. Solutions, minerals, and equilibria. San Francisco: Freeman Cooper. Genin, J-M. R., A. A.Olowe, Ph.Refait, and L.Simon. 1996. On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate or sulphate-containing green rust 2. Corrosion Science38:1751–62. Gettens, R. J.1963. The corrosion products of metal antiquities. In Annual report to the Trustees of the Smithsonian Institution for 1963. Washington, D. C.: Government Printing Office. 547–68. Goldschmidt, J.. 1953. A simplexity principle. Journal of Geology61:539–51. Gudas, J. P., and H. P.Hack. 1979. Sulfide-induced corrosion of copper-nickel alloys. Corrosion35:67–73. Hack, H. P., H.Shih, and H. W.Pickering, 1986. Role of the corrosion product film in the corrosion protection of Cu-Ni alloys in seawater. In Surfaces, inhibition, and passivationed.E.McCafferty and R. J.Broadd. Pennington, N. J.: Electrochemical Society. 355–67.
Heimann, R. B.1989. Assessing the technology of ancient pottery: The use of ceramic phase diagrams. Archaeomaterials31:123–48. Jambor, J. L.J. E.Dutrizac, A. C.Roberts, J. D.Grich, and J. T.Szymanski. 1996. Clinoatacamite, a new polymorph of Cu2(OH)3Cl, and its relationship to paratacamite and “anarakite.”Canadian Mineralogist34:61–72. Kato, C., and H. W.Pickering, 1984. A rotating disk study of the corrosion behavior of Cu-9.4Ni-1.7Fe alloy in air-saturated aqueous NaCl solution. Journal of the Electrochemical Society131:1219–24. King, F., D.LeNeveu, S.Ryan, and C.Litke, 1992. Predicting the long-term corrosion behaviour of copper nuclear fuel waste containers. In Life prediction of corroding structures, ed.R. N.Parkins. Houston, Tex.: National Association of Corrosion Engineers. 497–512. Kostov, I., and J.Minceva-Stefanova. 1981. Sulphide minerals. Sofia, Bulgaria: Publishing House of the Bulgarian Academy of Sciences. Kracek, F. C.1946. Phase relations in the system silver-sulfur and the transitions in silver sulfide. Transactions of the American Geophysical Union27:367–74. Lichtner, P. C., and M. S.Seth, 1996. User's manual for multiflo, parts 1 and 2. Center for Nuclear Waste Regulatory Analyses Report96–010. Little, B. J., P. A.Wagner, W. G.Characklis, and W.Lee. 1990. Microbial corrosion. In Biofilms, ed.W. G.Characklis and K. C.Marshall. New York: John Wiley and Sons. 635–70. Little, B., P.Wagner, and J.Jacobus. 1988. The impact of sulfate-reducing bacteria on welded copper-nickel seawater piping systems. Materials Performance27(8):57–61. Little, B., P.Wagner, J.Jacobus, and L.Janus. 1989. Evaluation of microbiologically induced corrosion in an estuary. Estuaries12(3):138–41. Livingston, R. A.1991. Influence of the environment on the Statue of Liberty. Environmental Science and Technology25:1400–08. Lucey, V. F.1967. Mechanism of pitting corrosion of copper in supply waters. British Corrosion Journal2:175–85. Macdonald, D. D., B. C.Syrett, and S. S.Wing. 1979. Corrosion of Cu-Ni alloys 706 and 715 in flowing sea water. Part 2, Effect of dissolved sulfide. Corrosion35:367–78. MacLeod, I. D., and C.Kenna. 1990. Degradation of archaeological timbers by pyrite: Oxidation of iron and sulphur species. Proceedings of the 4th ICOM-Group on Wet Archaeological Materials Conference, ed.PerHoffmann. Bremerhaven: Deutsches Schiffahrtsmuseum. 133–41. McNeil, M. B., J.Jones, and B. J.Little. 1991. Mineralogical fingerprints for corrosion processes induced by sulfate-reducing bacteria. Paper delivered at National Association of Corrosion Engineers International Corrosion/91, Houston, Tex. McNeil, M. B., and B. J.Little. 1990. Mackinawite formation during microbial corrosion. Corrosion46:599–600. McNeil, M. B., and B. J.Little. 1992. Corrosion mechanisms for copper and silver objects in near-surface environments. Journal of the American Institute for Conservation31:355–66. McNeil, M. B., and D. W.Mohr. 1992. Interpretation of bronze disease and related copper corrosion mechanisms in terms of log activity diagrams. In Materials problems in art and archaeology, vol. 2, ed.D. A.Scott. Pittsburgh: Materials Research Society. 1055–63.
McNeil, M. B., and D. W.Mohr. 1993. Formation of copper-iron sulfide minerals during corrosion of artifacts and implications for pseudogilding. Geoarchaeology8(1):23–33. Mond, L., and G.Cuboni. 1893. On the nature of antique bronze patina. Atti reale Accademia dei Lincei serie52:498–499. Mor, E. D., and A. M.Beccaria. 1975. Behaviour of copper in artificial seawater containing sulphides. British Corrosion Journal10:33–38. North, N. A., and I. D.MacLeod. 1986. Corrosion of metals. In Conservation of archaeological objects, ed.C.Pearson. London: Butterworths. 69–98. North, R. F., and M. J.Pryor. 1970. The influence of corrosion product structure on the corrosion rate of Cu-Ni alloys. Corrosion Science10:297–311. Ostwald, W.1906. Lehrbuch der Allgemeinen Chemie. 2d ed.Leipzig: W. Engelmann. Pfennig, N., F.Widdel, and H. G.Truper. 1981. The dissimulatory sulfate-reducing bacteria. In The prokaryotes: A handbook on habitats, ed.M. P.Starr et al. New York: Springer-Verlag. 926–40. Pollard, A. M., R. G.Thomas, and P. A.Williams. 1989. Synthesis and stabilities of basic copper. Part 2, Chlorides atacamite, paratacamite, and botallackite. Mineralogical Magazine53:557–63. Postgate, J. R.1979. The sulphate-reducing bacteria. Cambridge: Cambridge University Press. Pourbaix, M.1966. Atlas of electrochemical equilibria in aqueous solutions. Houston, Tex.: National Association of Corrosion Engineers. Putnis, A.1977. Electron diffraction study of phase transformations in copper sulfides. American Mineralogist62:107–14. Romanoff, M.1957. Underground corrosion. National Bureau of Standards Circular 579. Washington, D.C.: Government Printing Office. Rose, A. W.1969. Mobility of copper and other heavy metals in sedimentary environments. In Sediment hosted copper deposits, ed.R. W.Boyle, A. C.Brown, W.Jefferson, E. C.Jowett, and R. V.Kirkham, Geological Association of Canada special paper 36. Montreal: Geological Association of Canada. Roseboom, E. H.1966. An investigation of the system Cu-S and some natural copper sulfides between 25 degrees and 700 degrees. Economic Geology61:641–72. Rowlands, J. C.1965. Corrosion of tube and pipe alloys due to polluted seawater. Journal of Applied Chemistry15:57–63. Roy, R., A. J.Majumdar, and C. W.Hulbe. 1959. The Ag2S and Ag2Se transitions as geologic thermometers. Economic Geology54:1278–80. Scott, D. A.1990. Bronze disease: A review of some chemical problems and the role of relative humidity. Journal of the American Institute for Conservation29:193–206. Shcherbina, V. V.1978. The geochemistry of monovalent sulfides. Geokhimiya10:1444–51. Sinclair, J. D.1982. The tarnishing of silver by organic sulfur vapors. Journal of the Electrochemical Society129:33–40. Staffeldt, E. E., and D. A.Kohler. 1973. Assessment of corrosion products removed from “La Fortuna,” Punta del Mar. Venice: Petrolia e Ambiente. 163–70. Syrett, B. C.1977. Accelerated corrosion of copper in flowing pure water contaminated with oxygen and sulfide. Corrosion33:257–62.
Syrett, B. C.1980. The mechanism of accelerated corrosion of copper-nickel alloys in sulfide polluted seawater. Paper presented at National Association of Corrosion Engineers International Corrosion/80, Houston, Tex. Syrett, B. C.1981. The mechanism of accelerated corrosion of copper-nickel alloys in sulphide-polluted seawater. Corrosion Science21:187–209. Tribe, A.1998. Conservation report. In P. Clarke, The Roman watermills and settlement at Ickham, near Canterbury, Kent. Canterbury Archaeological Trust Occasional Paper. Canterbury: Canterbury Archaeological Trust. Trudinger, P. A., and D. J.Swaine. 1979. Biogeochemical cycling of mineral-forming elements. Amsterdam: Elsevier. Volpe, L., and P. J.Peterson. 1989. The atmospheric sulfidation of copper in a tubular corrosion reactor. Corrosion Science29:1179–86. Wagman, D. D., W. H.Evans, V. B.Parker, R. H.Schumm, I.Halow, S. M.Bailey, K. L.Churney, and R. L.Nuttall1982. The NBS tables of chemical thermodynamic properties: Selected values for inorganic and C1 and C2 organic substances in SI units. Journal of Physics and Chemistry Reference Data11 (suppl. 2). Walton, J., and M. B.McNeil. 1994. Coupled mass-transport and chemical-equilibrium modelling of bronze disease. In High level radioactive waste management. LaGrange Park, Ill.: American Nuclear Society. 1014–19. Wikjord, A. G., T. E.Rummery, F. E.Doern, and D. G.Owen. 1980. Corrosion and deposition during the exposure of carbon steel to hydrogen sulfide water solutions. Corrosion Science20:651–71. FURTHER READINGGeilmann, W.1956. Leaching of bronzes in sand deposits. Angewandte Chemie68:201–11. Gounot, A. M.1994. Microbial oxidation and reduction of manganese: Consequences in groundwater and applications. Federation of European Microbiological Societies Reviews14:339–50. AUTHOR INFORMATIONMICHAEL MCNEIL took B.A. and M.A. degrees from Rice University in chemistry and a Ph.D. in metallurgical engineering from the University of Missouri at Rolla. He did research in materials science at the Universities of Birmingham and Bristol (U.K.), Iowa State University, and Mississippi State University before joining the U.S. Civil Service as a materials scientist. His primary activities in recent years have been microbiological corrosion, particularly of submerged and buried objects, and grain boundary effects on metal degradation. At present he is employed in the Office of Research of the Nuclear Regulatory Commission. Dr. McNeil is also adjunct professor of materials science at the University of Toronto. BRENDA J. LITTLE, senior scientist for Marine Molecular Processes at the Naval Research Laboratory, has a Ph.D. from Tulane University in chemistry and a B.S. from Baylor University in biology and chemistry. Dr. Little has adjunct faculty positions at Montana State University and the University of Southern Mississippi. She is a member of the American Chemical Society, and the National Association of Corrosion Engineers (NACE International), Sigma Xi, and the Mississippi Academy of Sciences. She serves on the editorial board for Biofouling, a journal dealing with bioadhesion and biofilm research. She is the author of one book, 15 book chapters, and more than 80 peerreviewed journal articles.
 Section Index Section Index |