POLARIZED LIGHT MICROSCOPY IN CONSERVATION: A PERSONAL PERSPECTIVEWALTER C. McCRONE
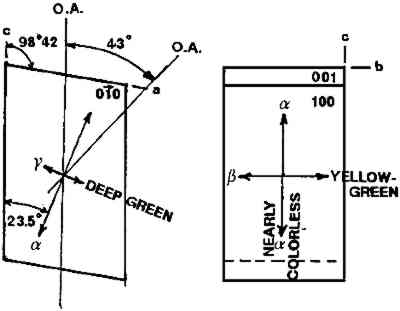
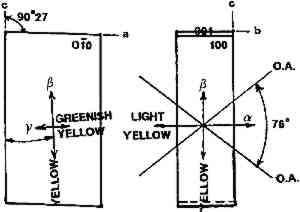
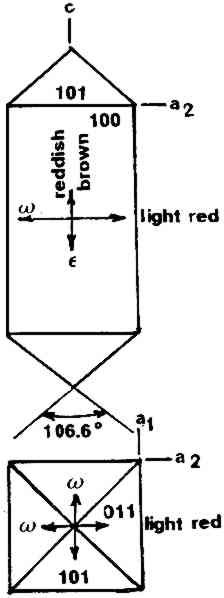
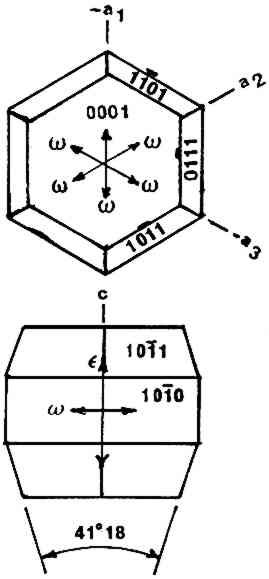
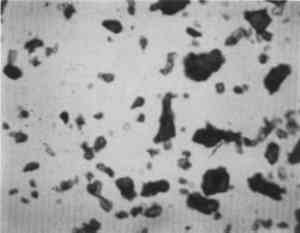
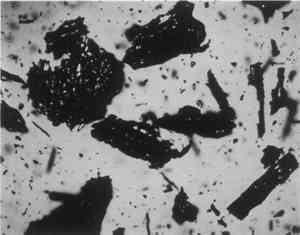
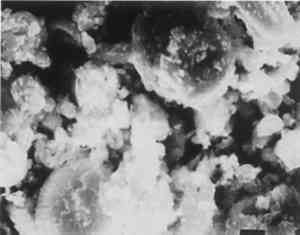
ABSTRACT—A conservator in any area of art or archaeology should know what materials he or she is dealing with. The microscopist quickly identifies metals, minerals, ceramics, vegetable and wood fibers, animal hairs (species), textile fibers, corrosion products, pigments, media, supports, dyes, inks, etc. Although many other useful microanalytical techniques are available, the only instrument capable of such complete characterization and identification for such a variety of diverse substances is the polarized light microscope. 1 INTRODUCTIONUntil the second half of this century the polarized light microscope held a dominant position in microanalysis. It was often the instrument of choice even when there was sufficient sample available for use of more macro methods of analysis (Schneider 1964; Benedetti-Pichler 1965). There were many university professors of polarized light microscopy (PLM) in both chemistry and geology departments, notably E. M. Chamot and C. W. Mason at Cornell, N. F. Witt and C. F. Poe at Colorado, and M. L. Willard at Penn State. Shortly after World War II, however, transmission electron microscopy and infrared absorption spectroscopy were finding a place in industry. These were soon followed by the scanning electron microscope and the electron and ion microprobe analyzers. On the assumption that these high technology instruments rendered the polarized light microscope (PLM) obsolete, nearly all colleges and universities dropped their microscopy courses, and many industries stored their light microscopes and reassigned their light microscopists. Only a very few individuals and industries maintained their expertise in this nearly abandoned field. Those microscopists who maintain their ability to apply PLM and its adjunct, optical crystallography, have continued because they are able to solve complex microanalytical problems quickly and with certainty. Unfortunately, nearly all have to operate in a world unaware and unappreciative of PLM. Often their results and conclusions are not accepted by the scientific world. A prime example is research on the Turin shround (McCrone 1990b). From the evidence of PLM, this venerated cloth is a beautiful painting: red ochre and vermilion pigments in a collagen tempera medium on a 14th-century canvas. There is absolutely no doubt concerning this conclusion, yet it has been accepted only by a few individuals knowledgeable about PLM or by others who were convinced only by the carbon date of 1325 � 65 years. Most others believe it to be the first-century shroud of Christ. Such reactions and receptions for microscopical results cause acute frustration among light microscopists. The Vinland map (McCrone 1988) is another example. We reported this to be a 20th-century effort. Observations with the stereomicroscope at the time of sampling the ink indicated the map had to be a forgery. The usual yellow stain that forms over time along ancient black ink lines had been inked on. This proved an intentional fraud. Later, we found the yellow “stain” contained a yellow titanium white (TiO2) in synthetic pigment size and form, but characteristic of titanium white pigments as produced during the early 1920s before all of the ilmenite iron was removed from the TiO2. It was therefore, yellow, and it was sold only for off-white paints. This negative finding was accepted until another laboratory (Cahill 1987) reported the TiO2 is present in far too low a percentage to account for the amount we reported in the yellow ink. However, this laboratory's high-tech cyclotron procedure required a large fraction of a milligram sample, in which it found 0.0062% TiO2. The yellow TiO2 is not, however, dispersed throughout the parchment sample but concentrated in the very thin yellow ink borders, where it constitutes up to 45% of the yellow ink. These findings illustrate the difference between ultramicroanalysis of a small, highly concentrated sample and a trace analysis of a large sample with a very low concentration of the component in question. Such differences are evident to microscopists. Because my microscopical results are rejected by art scholars, I have recently decided to refuse to analyze the pigments in any painting where the objective is authentication. The fact remains that PLM in the hands of trained microscopists solves many problems faced by art conservators quickly, confidently, and with a small capital investment. 2 USES OF MICROSCOPYMicroscopy is defined as the application of any enlarged-image process enabling visualization of objects nominally invisible to the unaided human eye. Individuals using the instruments capable of producing enlarged images of tiny objects are called microscopists. Those individuals, properly trained, are able to extend the observation and interpretation of large (macroscopic) objects down to subnanometer objects difficult to identify by any other means. The PLM, by producing enlarged images and by examining samples between polarizing filters, makes it possible to see, isolate, identify, measure, interpret, and evaluate microscopic objects (Chamot and Mason 1958; Hartshorne 1960). After microscopical enlargement, most tiny pigments, fibers, etc., are usually identified by a process all of us use thousands of times a day to recognize people, buildings, trees, books, cars, etc. Many microscopic substances enlarged 10-1000x are then recognized just as definitely, and just as rapidly, as we recognize macroscopic objects without magnification. There are look-alikes, however, that require even experienced microscopists to hesitate, consider the possibilities, and decide how to differentiate between them (e.g., bast fibers, starch grains, wood fibers, and some blue or yellow pigments). Then more definitive measurements must be made. Usually a few simple and rapid tests are all that is required to reduce the number of possibilities, most often, to one. These might include closer attention to shape characteristics, measurement of refractive index, melting point, difference between the refractive indices (birefringence), or orientation of the refractive indices relative to “crystallographic” directions in the particle or fiber. For example, corn and rice starch, otherwise identical, differ in size by a factor of 2 (10 μm and 5 μm diameters, respectively); alizarin has a refractive index higher than the standard refractive index liquid (nD = 1.66), while its look-alike, madder, is lower; whiting has one refractive index equal to nD = 1.66 and is rhomb-shaped, while other common carbonate minerals with identical shapes all have higher indices. Fortunately, each of the pigments, as well as other crystalline substances, have a unique set of physical measurements (McCrone et al. 1984) that are made by PLM to ensure identification. Figures 1–4 show some of the crystallographic data for four representative pigments. No other substance in the universe is likely to match any of these four sets of morphological and optical data.
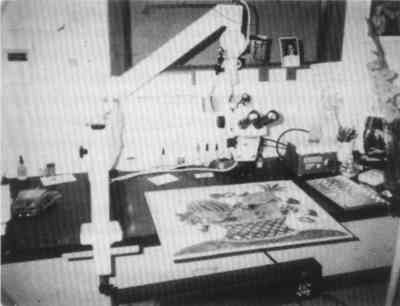
The PLM, unlike most other microanalytical tools, is equally applicable to organic, inorganic, biological, crystalline, or noncrystalline unknowns. It does, however, have the major disadvantage that a great deal of training and experience are necessary for best use. One might consider that becoming a competent microscopist is equivalent to learning to play a musical instrument well. After an introduction to the methods of microscopical analysis through formal instruction,1 these methods are best learned by actual observation and practice. The courses available (e.g., microchemical analysis, fiber and hair analyses, polarized light microscopy for the conservator, etc.) only tell what can be done and how to perform the techniques for proper use of the microscope and characterization of substances of interest. The student must observe, practice, and learn to use those techniques “on the job.” PLM data are far more definitive than the data obtained by SEM/EDS (x-ray microanalysis with the scanning electron microscope) data or even FTIR/microscopy (Fourier transform infrared absorption spectroscopy with microscope attachment), two widely used micro-analytical instruments. PLM not only identifies any pigments but also different forms of, say, lead white, lead oxides, or iron oxides. Lead white, the carbonate PbCO3 or the basic carbonate 2PbCO3�Pb(OH)2, are easily differentiated. There are several lead oxide pigments, including red lead, massicot, and litharge; each is distinctive in microscopical characteristics. These two other recently developed microanalytical instruments are, however, excellent supplements to the use of PLM. 3 INSTRUMENTATIONOf the wide array of microscopes available today, those most useful to the conservator are the stereomicroscope and the PLM. If one has to economize, the PLM in combination with a stereomicroscope (both for less than $10,000) are the best choices. These two microscopes are adequate for inspecting, sampling, microchemical analysis, hot stage microscopy, as well as crystallographic characterization. Perhaps the most generally useful (and used) microscope is the stereomicroscope, called by biologists a dissecting microscope. It is essential for sampling such materials as paint layers, patinas, corrosion products, paper, inks, textiles, and metals. It resides immediately adjacent to the PLM in most laboratories. Although limited in magnification and resolving power (about 150x and 2 μm), it provides an erect (rather than a reversed left to right) stereo image. It is very useful for the examination and sampling of leather, ropes, paintings, inks, stains, insects, and biological damage, as well as for the examination of brush marks on, or cross sections of, paint layers. The stereomicroscope is not generally used for determining chemical composition of such materials, although many such objects in the 0.05–1 mm size range are recognized by shape, size, and color. The 2–3 inch working distance below the stereomicroscope objectives allows plenty of room for dissecting needles and probes. The conservator's stereomicroscope might well be on an extension arm to make possible the examination and sampling of paintings (fig. 5).
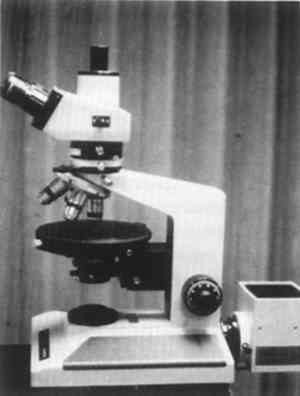
The conservator's PLM might also have an inclined binocular head for easier use (fig. 6), and a trinocular head will facilitate photomicrography or video microscopy. Not necessary are an automatic exposure camera, a heavy stand, confocal illumination, phase contrast, Nomarski, or Hoffman contrast accessories. A simple 35 mm or 4 � 5 Polaroid camera with an accessory exposure meter completes the major equipment needs of the conservation microscopist. A few books, reagents, solvents, microtools, refractive index liquids, slides, and coverslips (fig. 7) complete the basic microscopy laboratory.
If funds are available, two other very useful instruments are the SEM/EDS and the FTIR/microscope. If such instrumentation is unavailable internally, samples can be sent to a commercial laboratory. SEM/EDS is a method for elemental analysis of subnanogram samples suitable for the detection of most of the 104 chemical elements (at least sodium and above in the periodic table), but it is limited since it yields only an elemental analysis. Many combinations of pigments would yield similar EDS spectra, especially since the elements below sodium in the periodic table are “invisible” to EDS when using a detector with a beryllium window. For example, the lead oxides PbO and Pb3O4 would yield identical spectra—identical, as well, to the spectra of lead white and lead carbonate. SEM/EDS is most useful for differentiating between pigment look-alikes (e.g., ultramarine and smalt, hematite and vermilion), and an experienced PLM analyst is often relieved to have confirmation by SEM/EDS of such look-alikes in a given sample. Wavelength dispersive x-ray analysis by the far more expensive analytical transmission electron microscope or electron microprobe has a higher energy resolution and lower detection limits than EDS. The ion microscope (microprobe) covers all of the periodic table with subpicogram sensitivity. These latter instruments are quantitatively quite accurate. FTIR/microscopy is also an excellent supplement to PLM for the identification of pigments (Low and Baer 1977). It has the added advantage that it also characterizes media, varnishes, adhesives, and coatings. The FTIR/microscope yields an infrared absorption pattern for subnanogram samples. Like the SEM/EDS, however, it identifies parts of the molecules but, often, not how the parts are assembled. If the sample is a pure substance, pure only in the sense of a wax, varnish, paint medium, etc., the FTIR/microscope may well identify the sample as such and often even whether the wax is beeswax, camauba, etc. Other microanalytical instruments are only very occasionally useful to the conservator. While a few other analytical tools may identify some subnanogram samples, no instrument or technique other than PLM identifies all such particles, whether biological (e.g., species of pollen, wood, animal hair, etc.), crystalline or noncrystalline, organic or inorganic. Few analysts are able to isolate, clean, and mount a single subnanogram particle for x-ray diffraction analysis, SEM/EDS, or FTIR/microscopy. The PLM analyst does this routinely; still, it is not always necessary to separate individual particles from a matrix, as they are characterized and identified even in mixtures. Particles are visually separate, one from the other, in the microscope field of view. 4 TECHNIQUESSampling of art and archaeological subjects and paintings (McCrone 1982) is usually done with very fine needles (fig. 8) often made as needed by etching the ends of 0.5 mm tungsten wire (24 gauge) with sodium nitrite as described in the Particle Atlas(McCrone et al. 1992). A reasonable substitute may be made in a few minutes from a steel dissecting needle using very fine emery paper. The tip diameter viewed from the side should be 5–10 μm in order to handle tiny samples in that size range. Samples smaller than the resolution limit of the unaided human eye are less than 0.1 mm in size, but they often consist of hundreds of individual pigment particles. This sample size is more than adequate for characterization or identification of the pigment components by PLM.
Low magnification, say 10–50x with the stereomicroscope, aids in sampling paint surfaces, either ultramicro samples with the fine tungsten needle or small cross sections with a slightly coarser needle tip diameter or a fine scalpel. We make our own needles and scalpels. We prepare tiny scalpels by breaking 1–2 mm lengths of a fresh, two-edged safety razor using fine-tipped pliers. When necessary, we grind the pliers tips to about 1 mm2 gripping area. The small bits of razor blade, with only 2–5 mm of cutting edge, are placed in a vial for future use. When needed, a likely candidate is removed with fine-tipped forceps and oriented in a needle-holding chuck with the razor edge exposed and oriented at an appropriate angle (usually 30–60� from the length of the chuck). Tightened firmly, it furnishes a very sharp edge nicely oriented to scrape a surface or cut a thin section to mount for study and testing by stereomicroscope and PLM. The thin section is held in place on edge in a drop of thermoplastic Aroclor 5442 or embedded in polyester or epoxy in order to grind and polish a more formal surface through the various paint layers. Staining and other tests are performed directly on either type of section, and individual paint layers are sampled with a fine tungsten needle for study by PLM. Samples for identification are usually mounted dry under a coverslip taped onto one end of a microscope slide (McCrone 1982). Tinier portions of that sample are taken with the fine tungsten or steel needle tip and mounted in the center of the same slide using Aroclor, an ideal permanent mounting medium (McCrone 1984b). Any individual particle is removed later if necessary (McCrone 1984a) for characterization by other microanalytical methods (SEM/EDS, FTIR/microscopy, etc.). The PLM is used by mineralogists to identify rocks and minerals and by chemical microscopists to identify a wide variety of microscopic particles, for example, pollens and spores, manmade and natural fibers, minerals, crime lab trace evidence, industrial powders, metals, contaminants, and settled dust (McCrone et al. 1992). Thousands of tiny particles are identified by noting size, shape, color, transparency, surface topography, edge contrast, etc., almost at sight by a trained polarized light microscopist. Cotton (fig. 9) and hemp fibers (fig. 10) are each identified as such, at sight, as are softwood (fig. 11), and wool (fig. 12), as well as pigments such as gamboge (fig. 13), alizarin (fig. 14), vermilion (fig. 15), and charcoal (fig. 16). Fortunately, a polarized light microscopist becomes so familiar with the appearance of many substances that even with quick confirmatory tests and observations, recognition is nearly as rapid as identifying microscopes, telephones, cameras, or an automobile macroscopically. The quick confirmatory tests are as simple as crossing the polars, focusing up and down, or rotating the microscope stage. With very little more time, the species of pollens, animal hairs, or vegetable fibers, composition of gilt, or the identification of paint media is determined by the PLM microscopist experienced in those areas.
For those particles not identified at sight, PLM has an arsenal of tests designed to produce sufficient additional data to help in identification (McCrone et al. 1984). First, the refractive index relative to the mounting media is quickly estimated qualitatively. The readings, based on darkness of the particle boundaries (contrast) and the Becke line behavior on focusing up and down through best focus might be nearly equal, higher, lower, much higher, or much lower. Rotating the stage may yield a similar observation for a different orientation of the particle relative to the vibration direction of the polarized light. Next, crossing the polars causes some substances to disappear against the resulting black background (e.g., cubic crystals and glasses). Other particles appear bright, and the colors (and their intensity) correspond to the difference between the two refractive indices shown by those particles; this is estimated (e.g., small, medium, or large) or measured quantitatively. On rotation of the stage, all such colored particles disappear (show extinction) every 90�. The particle orientation at extinction is characteristic and helps as an additional identifying characteristic. All these observations are very rapidly carried out. There are others, less rapidly performed but increasingly definitive. These include actual measurement of one or more refractive indices by immersion in standard Cargille refractive index liquids to find a match; addition of a compensator to determine the orientation of the high refractive index; measurement of the extinction angle (the angle between extinction and a prominent particle shape direction); and measurements on the interference figure. The latter might well be described as a “fingerprint” for a crystalline substance. A valuable group of quantitative and qualitative measurements are made on such figures. All these observations and measurements usually consume 15–20 minutes. In addition, one may elect to measure thermal properties using a hot stage or to perform microchemical tests in an aqueous drop using standard reagents for inorganic ions or for organic compounds. A series of staining tests on single particles of a starch grain or a crystal of Prussian blue are examples of this quick test procedure. Seldom is it necessary to perform more than a few of these tests given the knowledge that the particles are one of about 10 pigments of a given color, one of the small number of paint media, or one of the common starch grains or bast fibers. Still, a microscopist should ideally use the best and surest analytical tool to solve the tougher problems. One such problem is the differentiation of cadmium red, red lead, vermilion, hematite, and red ochre. All are finely divided red pigments, and all have refractive indices higher than any Cargille refractive index liquid. Most pigments are identifiable at sight, but an elemental analysis by SEM/EDS helps solve problems like the identification of these red pigments or other look-alikes. Vermilion shows mercury and sulfur, red lead shows lead, iron earth pigments show iron and (with the umbers) manganese, cadmium red shows cadmium, sulfur, and/or selenium. These microchemical tests can be made in single-drop tests on a microscope slide, but, when available, most microscopists prefer to use SEM/EDS. This technique also helps to differentiate among ultramarine, smalt, and Prussian blue or among lead-tin yellow, zinc yellow, and Naples yellow, and other such look-alike problems. Some microscopists even add the whites (lead, zinc, and titanium) to this list. With care and a good microscopical background, however, all can be differentiated by PLM. It is also possible to carry out chemical reactions as another means of identifying pigments, corrosion products, minerals, or metals. These tests generally involve single-drop volumes of test substance and reagent resulting in precipitation of characteristic crystal formations, performed on tiny subnanogram samples when necessary (McCrone 1986). Single particles of most pigments are identified by microchemical tests using PLM. Likewise, single fibers and very short lengths thereof are uniquely characterized and identified microscopically by their optical properties. Many staining reactions as well as ultraviolet fluorescence are also adaptable to observation with PLM. In sum, the combination of PLM, FTIR/microscopy, and SEM/EDS effectively and efficiently solves 99% of the problems we face. I would have to add that a trained polarized light microscopist could likely solve more of the conservator's problems than both of the other two together, but having all three facilitates the solution of many problems. 5 APPLICATIONSThe conservator-microscopist can study the identity, composition, or condition of objects in museum or other collections, including textiles, manuscripts, paper, leather, wood, metals, ceramics, stone, paintings—in short, any artistic or archaeological object. One significant application of PLM is for the characterization of a painting relative to the authenticity of its attribution to a particular time or painter. The pigments used in paintings all have a time of first use, although for some the date is shrouded in antiquity. Since about 1300, however, historical records and the finding of specific pigments in paintings of known date have fixed a list of dates of first use for most post-A.D. 1300 pigments (McCrone 1987). Some pigment compositions have multiple dates because they appear in different crystal forms (polymorphs) at different times or in different combinations. Titanium white (table 1) is an excellent example (McCrone 1990a). McCrone (1990a) also describes how PLM can identify rutile and anatase as such. TABLE 1 HISTORY OF USE OF TITANIUM WHITE PIGMENT Whiting and vermilion present similar situations. Whiting in the crystal form of rhombohedral calcite occurs in nature as the limestone mineral (fig. 17), as the skeletal remains of marine plankton (a coccolithophore) found in chalk (in the cliffs of Dover, for example) (fig. 18), and as a synthetic product dating only from the early 1800s (fig. 19). Vermilion also occurs in three forms: the mineral cinnabar, a dry process vermilion first produced by alchemists about A.D. 800, and a wet process form first synthesized as a pigment about 1700. I have observed the A.D. 800 vermilion in only a few medieval paintings, but notably in the Turin shroud blood-image areas (McCrone 1990b).
The condition of medieval pigments compared with their modern counterparts is also helpful in dating paintings. In general, pigments today are purer and more finely divided. Earth pigments, especially the iron earth pigments, tend to contain more quartz, limestone, and feldspar contaminants in earlier paintings. The synthetic equivalents of the earth pigments are, of course, free of extraneous mineral impurities. Lead white in the carbonate form, PbCO3, is hexagonal, as is the basic carbonate 2PbCO3�Pb(OH)2, but the latter is usually produced as flat plates, whereas the carbonate is elongated in the undc` axis direction. I have seen the latter in only three paintings: two paintings known to be by Edouard Manet and another painting also very likely a Manet (McCrone 1987). This situation invited a closer study of these elongated crystals of lead white in the hope of supporting, if not proving, the attribution to Manet. Two individual crystals each were isolated from the Infanta Margarita and Ballet Espagnol and checked for trace elements. Some of the data are shown in table 2. TABLE 2 COMPOSITION OF FOUR LEAD WHITE PIGMENT PARTICLES The Infanta data are from the likely Manet painting and the Ballet is from a known Manet painting in the Phillips Collection in Washington, D.C. The data for the first three crystals listed are very close. The fourth set of data differ especially for aluminum, magnesium, and silicon—common elements everywhere. I assume I did not clean that particular subnanogram crystal as well as I cleaned the other three; the largest was about 2 � 2 � 10 μm. In any case, the data in table 2 indicate that those crystals had a common history. I concluded that they could not have been more similar if they had been squeezed from the same tube of paint (McCrone 1987). My final conclusion was that the Infanta was very likely the long-lost Manet copy of a portion of a Vel�zquez painting in the Louvre; the conclusion of one “scholar” was that perhaps someone borrowed Manet's paints one day and produced the Infanta. This is another experience that convinced me not to spend any more time trying to authenticate works of art. Fortunately, there are many interesting problems for the conservator. For example, environmental effects can be evaluated and elucidated by microscopy. Louis Pomerantz once brought to me a painting by Fernand L�ger owned by a Chicago collector. The blue paint layers persisted in developing a white particulate efflorescence. A few crystals removed from the surface were recrystallized from a small drop of water on a microscope slide. I recognized the well-formed crystals immediately because they happened to be borax, a compound we use in many of our PLM courses as an example of a monoclinic crystal. It is a hygroscopic substance, dissolving in condensed moisture from your breath or even in high humidity atmospheres. The efflorescence that appeared on the blue paint surface was a result of moisture drawn from humid air migrating into the paint layer, which contained borax. The borax dissolved, followed by migration of the resulting borax solution to the paint surface, where it crystallized during dry periods. Katherine Kuh, then at the Art Institute in Chicago, suggested that L�ger, a potter as well as an artist, probably ran out of his favorite blue pigment and substituted a blue pottery glaze that contained borax as a flux. Another more serious environmental problem occurs in sulfuric acid–laden city atmospheres, polluted by the burning of fossil fuels. Sulfuric acid, in contact with paint layers that have acid-soluble and reactive pigments, such as zinc white, reacts to form sulfates. The crystals of zinc sulfate, identified by PLM, apparently result from zinc white reacting with sulfuric acid in city atmospheres. They are hydrated and thus occupy much more space than the original zinc oxide. The increased volume of the zinc sulfate formed during this reaction exapands and breaches the paint layer surface. If present in more than trace amounts, this expansion literally destroys that area of the painting. The expansion within the paint layer quickly breaks openings in the paint surface, facilitating an increasing rate of disintegration. Obviously, such conditions must be watched carefully, and, when they are first detected, heroic efforts must often be taken to prevent further damage. 6 CONCLUSIONSThe advantages of microscopy are many. It is a direct method. EDS analysis tells us what elements are present but not what the compound is—for example, that calcium is present (could be whiting, gypsum, or anhydrite), or iron (could be Prussian blue or any iron earth pigment), or copper (could be malachite, azurite, copper resinate, verdigris, or blue verditer). The situation is little better if we find by FTIR a C-H stretching frequency and a carbonyl group. Yet, again, I prefer to have and to use all three instruments. SEM/EDS quickly decides between vermilion and hematite, smalt and ultramarine; the FTIR/microscope is a quick and sure way to identify adhesives, varnishes, and media. With this arsenal of instruments—the polarized light microscope supplemented as necessary with SEM/EDS and the FTIR/microscope—there are few analytical problems of the conservator that cannot be solved. NOTES1. Instruction in the use of the PLM for the conservator is available at graduate programs in art conservation and at independent facilities such as the McCrone Research Institute. REFERENCESBenedetti-Pichler, A.1965. Identification of materials. Vienna: Springer. Cahill, T. A.1987. The Vinland Map, revisited. Analytical Chemistry50:829–3. Chamot, E. M. and C. W.Mason. 1958. Handbook of chemical microscopy, vol. 1. New York: Wiley. Hartshorne, N. H.1960. Crystals and the polarizing microscope. London: Arnold. Low, M. J. D. and N. S.Baer. 1977. Application of infrared Fourier transform spectroscopy to problems in conservation. Part 1. General principles. Studies in Conservation22:116–28. McCrone, W. C.1982. The microscopical identification of artists' pigments. Journal of the International Institute for Conservation—Canadian Group7(1–2):11–34. McCrone, W. C.1984a. Tricks of the trade: Rescuing a particle from an Aroclor mount. The Microscope32:148–49. McCrone, W. C.1984b. Use of Aroclors in microscopy. The Microscope32:277–88. McCrone, W. C.1986. Solubility, recrystalliztion and microchemical tests on nanogram single particles. The Microscope34:107–18. McCrone, W. C.1987. Authenticity study of a possible Manet painting. The Microscope35:173–95. McCrone, W. C.1988. The Vinland map. Analytical Chemistry60:1009–18. McCrone, W. C.1990a. 1500 forgeries. The Microscope38:289–98. McCrone, W. C.1990b. The Turin shroud: Blood or artist's pigment. Accounts of Chemical Research23:77–78. McCrone, W. C., J. G.Delly, et al. 1992. Particle atlas: Electronic edition. Hayward, Calif.: Micro-Dataware. Print edition 1974–1980. McCrone, W. C., L. B.McCrone and J. G.Delly. 1984. Polarized light microscopy. Chicago: McCrone Research Institute. Schneider, F. L.1964. Qualitative organic microanalysis. New York: Academic Press. SOURCES OF MATERIALSEquipment necessary for a conservator's PLM laboratoryMcCrone Accessories and Components 850 Pasquinelli Dr., Westmont, Ill. 60559 Commercial laboratories include:Charles Evans and Associates, 301 Chesapeake Dr., Redwood City, Calif. 94063 McCrone Group, 850 Pasquinelli Dr., Westmont, Ill. 60559 MVA, 5500 Oakbrook Parkway, Suite 200, Norcross, Ga. 30093 R. J. Lee Group, 350 Hockberg Rd., Monroeville, Pa. 15146 Structure Probe, P.O. Box 656, West Chester, Pa. 19381 Cargille refractive index liquids:R. P. Cargille Laboratories, 55 Commerce Rd., Cedar Grove, N.J. 07009 KAP extended arm stereomicroscope:Schleuter Instrument Corporation, Twin Lakes Technological Park, 4699 Nautilus Court, Suite 105, Boulder, Colo. 80301 AUTHOR INFORMATIONWALTER C. MCCRONE received B. chem. and Ph.D. degrees from Cornell University. He worked at the Illionois Institute of Technology Research for 12 years before starting his own research laboratory, McCrone Associates, in 1956. His special interests have always been chemical microscopy, crystallography, and ultramicroanalysis. He has more than 350 technical publications, including 12 books. In 1960 he started the McCrone Research Institute, devoted to teaching and fundamental research. He is editor and publisher of the international applied journal of microscopy, The Microscope. He is an Honorary Member of the American Institute for Conservation, a Fellow of the International Institute for Conservation and the American Academy of Forensic Sciences, an Honorary Fellow of the Royal Microscopical Society, and a member of many other societies. He has received a number of awards: Benedetti-Pichler (1970), Ernst Abbe (1977), Anachem (1981), and Criminalist of the Year (1985), among others. Address: McCrone Research Institute, 2820 S. Michigan Ave., Chicago, Ill. 60616–3292.
 Section Index Section Index |