CORROSION MECHANISMS FOR COPPER AND SILVER OBJECTS IN NEAR-SURFACE ENVIRONMENTSMICHAEL B. McNEIL, & BRENDA J. LITTLE
ABSTRACT—The morphology and mineralogy of corrosion products found on copper and silver artifacts recovered from shallow land or water burial are reviewed. Stability diagrams are used to interpret mechanisms for corrosion phenomena with an emphasis on identification of microbiological action. 1 INTRODUCTIONObjects made of silver and copper alloys have frequently been recovered from shallow land burial or shallow seas. Corrosion products may contain information as to the original composition of an alloy and the conditions to which it has been subjected. Extraction of information is facilitated by using stability diagrams for interpreting archaeometallurgical corrosion data, especially in cases where microbiological action has resulted in corrosion phenomena that are otherwise difficult to interpret. 2 STABILITY DIAGRAMSA fundamental tool for interpreting chemical phenomena is the stability diagram (Pourbaix 1966; Heimann 1989; Garrels and Christ 1965), which shows regions of thermodynamic stability for specified phases under particular restrictions. These diagrams—often called Pourbaix, phase, or pH-potential diagrams—are presented at fixed temperature and chemical conditions with axes of pH and Eh. pH is the negative logarithm of the effective hydrogen ion concentration used to express both acidity and alkalinity. Eh is a measure of the oxidation or reduction potential of the system. Low Eh values correspond to reducing conditions and high Eh values to oxidizing conditions.
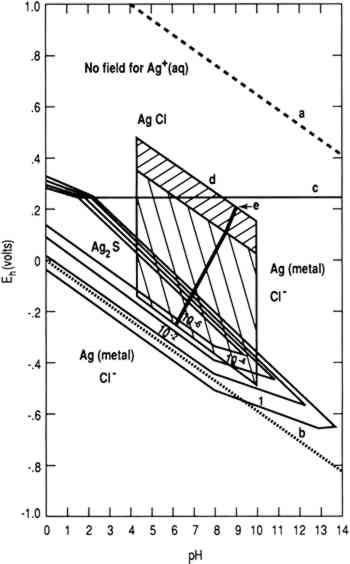
Figure 1 is a stability diagram for a silver-water-chloride-sulfur system constructed using thermodynamic data (Wagman et al. 1982). Line (a) is the oxygen line above which water is thermodynamically unstable
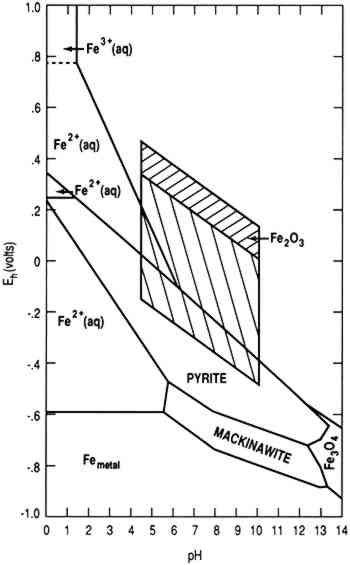
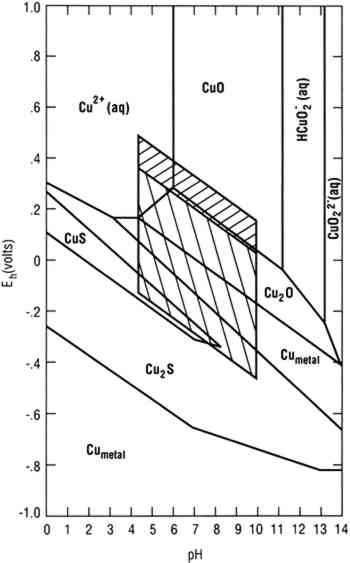
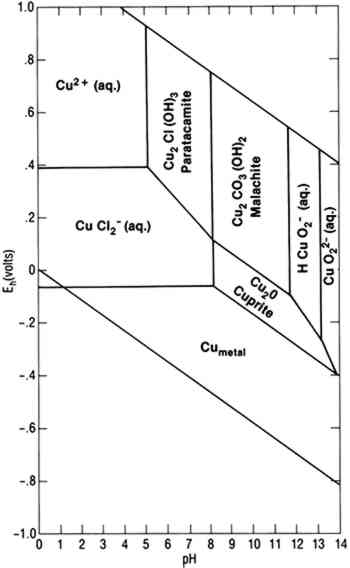
To achieve conditions at the surface of an artifact other than those defined by the two parallelograms requires the presence of a biofilm on the surface capable of maintaining conditions radically different from those in the bulk environment. Figure 1 indicates the possible thermodynamically stable phases for silver equilibrated with varying total sulfur compositions in 0.46 Molar NaCl (typical for seawater). It is assumed that reduced sulfur species (S−2, HS−, H2S) are in equilibrium. The area of the diagram above line (c) is the stability region for cerargyrite. Below line (c) metallic silver is stable, except for the wedge-shaped areas pointing down and to the right indicating regions of stability for monoclinic acanthite (Ag2S). Figure 2 is a similar diagram for an iron-water-reduced sulfur system with lines for 10−6 M dissolved iron and 10−2 M total reduced sulfur. Figure 3 is a stability diagram for copper and its minerals drawn for 10−6 M total dissolved copper and 10−2 M total reduced sulfur. Figure 4 is a stability diagram for copper that represents the condition when no sulfur is present and the electrolyte has the same chloride concentration as seawater and is in carbonate equilibrium with air. Total carbonate content (CO3−2, HCO3−, H2CO3) is assumed to be in equilibrium with air. Stability regions for red cuprite (Cu2O), malachite (Cu2(OH)2CO3), and paratacamite (Cu2(OH)3Cl) are shown. Thermodynamic data (excluding malachite) are from Wagman et al. (1982); malachite data are from Woods and Garrels (1966). Azurite (Cu3(OH)2(CO3)2) and georgite (Cu5(CO3)3(OH)4�6H2O) are not included. There are no thermodynamic data for georgite, and azurite has no stability field in water having carbonate content in equilibrium with air.
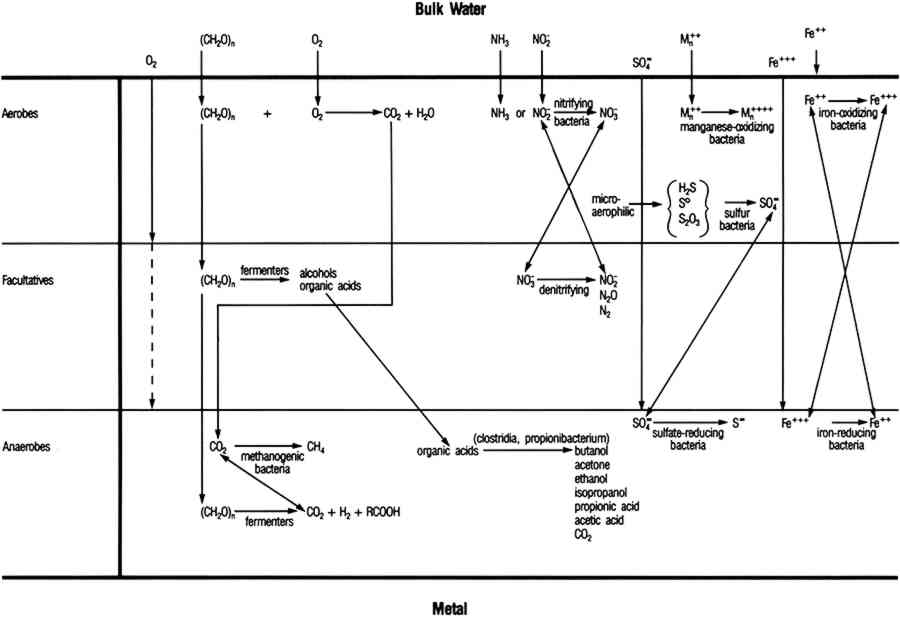
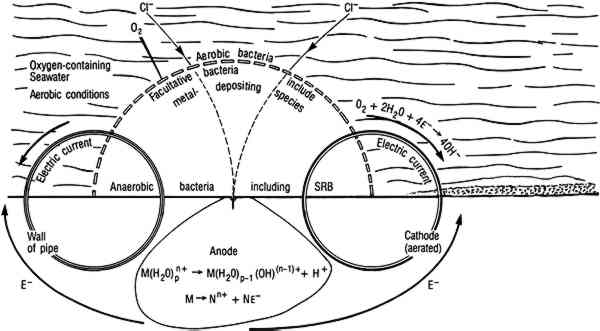
Stability diagrams can be used to analyze corrosion processes in the following way: A uniform coating of a particular mineral on the surface of an artifact indicates that surface conditions were such that the corrosion product was thermodynamically stable with respect to the metal and probably to other possible corrosion products. For example, some mechanistic information can be derived from the observation that the surface of a copper artifact was coated with azurite rather than malachite. Azurite is less stable than malachite in waters where the carbonate content is controlled by equilibrium with air. Azurite indicates that the artifact corroded Similarly, the formation of chalcocite (Cu2S) rather than covellite (CuS) in the presence of reduced sulfides provides evidence that surface conditions were such that chalcocite was stable. Conditions within a biofilm may be such that covellite is the stable product, but chalcocite may be detected in an x-ray diffraction pattern because it is present as an underlayer between the covellite and the copper alloy. Mass transport can lead to partial sulfidation of copper to chalcocite, then to covellite. Copper artifacts corroded in the absence of sulfides or sulfate-reducing bacteria (SRB) generally have a red cuprite layer under malachite or paratacamite. Equilibrium with the aqueous phase is limited to the outermost layer unless the corrosion product is porous. Based on the assumption that conditions are in local equilibrium, knowledge of electrolyte chemistry on the microscopic scale must be used in the analyses of corrosion product mineralogy. Two-dimensional presentations of phase equilibria controlled by two independent variables are adequate for corrosion processes influenced by redox behavior. Redox equilibria are not critical in determining corrosion processes in some cases, and more complex diagrams are required. For example, copper corrosion associated with simultaneous formation of cuprite and nantokite (CuCl) under a coating of cuprite is characteristic of both bronze disease (Scott 1990a) and type 2 pitting (Lucey 1967; Sato 1982). In other cases, corrosion products are divalent copper minerals (generally paratacamite in salt water and malachite or brochantite (Cu4SO4(OH)6) in fresh water). The corrosion chemistry is determined by the phase relationships of a number of cupric minerals. To present the phase equilibria in two-dimensional diagrams, it is necessary to construct projections of phase stability regions on planes perpendicular to the pH and Eh axes in four-dimensional figures with coordinate axes of pH, Eh, chloride activity, and copper activity. Explanations for using four-dimensional diagrams can be found in Zen (1966). Four-dimensional phase diagrams have also been used to interpret corrosion phenomena in bronze disease (Mohr and McNeil 1992; Scott 1990a) and type 1 pitting in copper. 3 MICROBIOLOGICALLY INFLUENCED CORROSIONIn natural environments bacteria attach to solids, including metals, where they colonize the surface and produce biofilms. Micro-organisms within the biofilm are capable of maintaining an environment at the surface of the artifact that is radically different from the bulk in terms of pH, dissolved oxygen, and other organic and inorganic species (fig. 5). In some cases, these interfacial conditions cannot be maintained in the bulk medium at room temperature near atmospheric pressure. For example, obligate anaerobic SRB that can only grow in anaerobic environments are routinely isolated from surfaces in oxygenated environments. SRB are a diverse group of bacteria that can be isolated from many environments, but their principal habitats are soil, sediments, and seawater (Miller and Tiller 1970; Battersby 1988). In natural environments SRB grow in consortia with oxygen-using microorganisms that can deplete the local environment, driving the conditions at the biofilm surface interface toward more negative Eh values and providing conditions for SRB growth. SRB are frequently isolated in association with tubercles formed by metal-depositing bacteria (Pope 1986). Within biofilms and tubercles SRB produce high concentrations of
4 SILVER CORROSIONAn example of silver corrosion is conversion of silver to cerargyrite, as indicated above line (c) in figure 1. The region between the diagonal lines that bound the upper hatched region approximates the effective oxidizing behavior of near-surface, fully aerated seawater (Garrels and Christ 1965). Most shallow sea chemistries fall into this region. Conditions are near this region in shallow land burials where the major source of groundwater is rain or surface water percolating through soils. Deep groundwaters and waters from coal mines may not fit this particular model (Garrels and Christ 1965). Cerargyrite is stable in seawater and chloride-rich shallow land burial. Silver and its alloys are subject to corrosion by reduced sulfur species. In air (e.g., in a museum) H2S can result from biodegradation of polymeric materials that contain sulfur, producing monoclinic acanthite (Banister 1952; Bauer 1988). In marine environments silver objects corrode under the influence of SRB to produce acanthite and cerargyrite. The formation of body-centered cubic argentite (Ag2S) has been reported (North and MacLeod 1986). Abiotic corrosion of pure silver in water that contains dissolved sulfides produces acanthite with traces of argentite (Campbell et al. 1982). The formation of argentite as a corrosion product is noteworthy because naturally occurring mineral argentite has not been reported in the biosphere and artificial argentite made with pure silver cannot be quenched to room There are three polymorphs of Ag2S. Monoclinic acanthite is stable up to 176�C (Kracek 1946). Body-centered cubic argentite is stable from 176�C to a temperature between 586�C and 622�C (Kracek 1946), above which the stable form is a face-centered cubic polymorph (Djurle 1958; Barton 1980). The high-temperature polymorph has never been observed in corrosion. Reactions interconverting Ag2S polymorphs are very fast. When acanthite forms in abiotic experiments, it does so in extremely small quantities as a surface phase (Campbell et al. 1982). The laboratory database on sulfides and their reactions with silver can be summarized as follows: (1) corrosion of silver by reduced sulfides, whether H2S (Sinclair 1982; Volpe and Peterson 1989) or organic sulfides (Sinclair 1982), produces acanthite; (2) carbon disulfide does not produce corrosion; (3) the corrosivity of organic sulfides appears to be controlled by transport mechanisms and thus by vapor pressures; and (4) the rate of sulfidation is strongly affected by NH3 and by alloying silver with iron (Biestek and Drys 1987). Abiotic aqueous corrosion of silver in the presence of reduced sulfur species produces acanthite in bulk (Birss and Wright The conversion of macroscopic quantities of silver to any sulfide in the biosphere requires the action of SRB, since the total reduced sulfide content of seawater and most groundwaters with pH below 9 is small. Except for the action of SRB, the conversion of sulfate to reduced sulfide requires geological time. Mor and Beccaria (1975) estimate zero sulfide concentration in Mediterranean seawaters having a typical sulfate concentration of 0.2 g/l and an SRB population of 10–102 per ml. These observations support the hypothesis that formation of argentite is limited to precipitation of silver in the presence of copper ions by a reduced sulfur species. This theory is consistent with the observation that argentite corrosion products are sometimes accompanied by jalpaite (Ag3CuS2) (North and MacLeod 1986). Argentite formed from pure silver is unstable at room temperature, yet there are two reasons why argentite should precipitate during the corrosion of archaeological objects. Argentite containing several percent copper is usually associated with jewelry and coinage. Unlike acanthite, it can accommodate nearly 30% copper in its lattice (Shcherbina 1978). The phenomenon is parallel to the production of akageneite (βFeOOH) rather than goethite (αFeOOH) in the corrosion of meteorites. The formation of akageneite is attributed to its ability to accommodate significant Cl−, while the goethite lattice can accommodate little or none (Buchwald 1977; Buchwald and Clarke 1989). Precipitation of a mineral from an impure environment favors a high-entropy, low-packing fraction crystal structure capable of accommodating impurity atoms (Goldschmidt 1953). Argentite formation occurs when an object made of a silver-copper alloy is in a water-saturated deposit that contains SRB capable of maintaining reducing conditions and bacteria (perhaps ammonia producers) capable of oxidizing and thereby solubilizing both silver and copper atoms. A layer of sand or soil restricts the ability of the metal ions to escape the biofilm, so that concentrations of copper and silver ions within it rise to levels that cause precipitation. The precipitation of argentite is favored for the reasons given above. Jalpaite forms in regions where the copper concentration is high enough to favor this mineral. In the absence of thermodynamic data the specific concentration cannot be quantified. Argentite and jalpaite could, in principle, be stabilized when formed by corrosion of pure silver in a copper-rich environment (such as that associated with a mixture of silver and copper coins), but for practical purposes the presence of either argentite or jalpaite implies that the silver artifact originally contained significant copper. 5 COPPER CORROSIONArtifacts are often made of copper containing several percent of alloying elements. Corrosion of such alloys is complex due to the large number of corrosion products that can form in the biosphere. The following discussion is limited to copper objects that corrode in water sufficiently oxidizing and near neutrality to permit the formation of cuprite in the absence of SRB. This limitation excludes reducing environments in which copper does not corrode and acid environments in which copper dissolves. These restrictions eliminate consideration of corrosion in peat bogs, some hot springs, and ocean bottoms off the continental shelf. The first product of copper corrosion, cuprite, forms epitaxially as a product of the direct reaction of copper with dissolved O2 or with water molecules (North and Pryor 1970). Cuprite has a high electrical conductivity and permits transport of copper ions (North and Corrosion of copper-nickel alloys in saline environments is much less rapid than corrosion of pure copper. Swartzendruber et al. (1973) proposed that this corrosion resistance was due to changes in the crystal chemistry of the base metal. This theory is no longer accepted. North and Pryor (1970) maintained that the improved corrosion resistance of copper-nickel alloys was due to changes in the mass transport characteristics of the oxide as a result of the addition of nickel. A comprehensive review of recent experimental work (Hack et al. 1986) indicates that, under hydrodynamic conditions that prevent accumulation of porous outer layers of hydroxychloride or hydroxycarbonate corrosion products, the critical effect of the alloying elements is to alter the cathodic oxygen reduction. The mineralogy of the corrosion products observed during long-term corrosion of copper alloys is different from that commonly found on pure copper. Nickel compounds are observed in an adherent corrosion product layer on copper-nickel alloys. Copper-tin alloys tend to form layered structures with the characteristics of Liesegang structures (Scott 1985). Zinc tends to be selectively removed from copper-zinc alloys, producing a spongy copper material (Walker 1977), although the incorporation of zinc in corrosion products to produce rosasite ((Cu,Zn)2CO3(OH)2) is known (Gettens 1963b). Dealloying of the copper from tin bronzes has been reviewed by Geilmann (1956). Partitioning of alloying elements between remaining metal ions in the corrosion product and the electrolyte has received little attention (Zolotarev et al. 1987). Review of the corrosion processes of bronzes and brasses might lead to reconsideration of their original alloy chemistries. If conditions at a copper surface permit precipitation of nantokite under the cuprite layer, the alloy becomes vulnerable to bronze disease (Scott 1990a) or type 1 pitting corrosion (Lucey 1967), depending on mass transport conditions. A biofilm containing acid-producing bacteria could help produce the requisite conditions by increasing acidity in anodic areas and reducing copper ion transport from the surface, resulting in a higher local copper concentration (Mohr and McNeil 1992). While nantokite-based corrosion can also occur by nonbiological paths, the nonisometric morphology frequently observed on precipitated cuprite crystals suggests that microbiological poisoning of growth planes is a factor. The presence of alloying elements does not protect against the formation of nantokite In addition to nantokite, melanothallite (Cu2OCl2), eriochalcite (CuCl2�H2O), and other copper chlorides and hydroxychlorides may produce bronze disease-type reactions. The failure to identify bronze disease precursors other than nantokite may be due to their rarity as corrosion products, to kinetic or thermodynamic factors, or to a lack of adequate experimental study. Copper alloys (except possibly arsenical bronzes) are also subject to sulfide-induced corrosion by SRB within a biofilm. Under these circumstances chalcocite forms easily, covellite less easily (fig. 3)(Syrett 1977, 1981; Kato et al. 1984). The corrosion layer may contain other sulfides buried under other corrosion products (Mor and Beccaria 1975; North and MacLeod 1986). Nonetheless, the poor adherence and mechanical properties of the sulfides make these layers nonprotective. The presence of iron ions in solution can lead to the formation of a layer of chalcopyrite (CuFeS2) (Daubr�e 1862; de Gouvernain 1875; Duncan and Ganiaris 1987). The circumstances under which chalcocite and covellite form and their relation to other sulfide corrosion products such as djurleite (Cu1.96S), and digenite (Cu5S9) have been addressed by McNeil et al. (1991). Under some conditions copper phosphate corrosion products can form. Libethenite (Cu2PO4OH) and sampleite ((Na,K) Ca Cu5 PO4Cl�5H2O) have been identified by Geilmann (1956) and Fabrizi et al. (1989), respectively. Some research on the consequences of deliberately phosphating copper surfaces has been reported (Reiber 1989), but basic data on copper phosphate minerals are lacking. 6 CONCLUSIONSCorrosion products associated with artifacts can provide information about original composition and the environment in which they were preserved. Two- and four-dimensional phase diagrams contribute to an understanding of the chemical-microbiological and historic environments. In cases where Eh and pH are the critical independent variables and where changes in concentrations of other ions do not fundamentally change the corrosion process, two-dimensional phase diagrams can be used. In more complex processes where corrosion products have finite solubility, it is necessary to construct four-dimensional figures with two-dimensional projections. Four-dimensional diagrams and projections have not until recently been used in corrosion science. ACKNOWLEDGEMENTSThe authors acknowledge Michael Marron of the Office of Naval Research, Code 1141MB, for financial support. David Mohr, Naval Coastal Systems Center, and Patricia Wagner, Naval Oceanographic and Atmospheric Research Laboratory, provided technical assistance. NOARL Contribution Number JA 333:081:90. REFERENCESBanister, F. A.1952. An unusual synthesis of acanthite crystals. Paper presented at the Mineralogical Society of London Meeting. Documented in the Ford-Fleischer files, U.S. Geological Survey, Reston, VA. Battersby, B. L.1988. Sulphate-reducing bacteria. In Methods of aquatic bacteriology, ed.B.Austin. New York: John Wiley and Sons. 269–99.
Barton, M. D.1980. The Ag-Au-S system. Economic Geology75:303–16. Bauer, R.1988. Sulfide corrosion of silver contacts during satellite storage. U.S. Air Force report SD-TR-88-53/AD-1196 217 (unclassified). Biestek, T., and M.Drys. 1987. Corrosion products forming on silver in various corrosive environments (in Polish). Powlocki Ochronne (Warsaw) 9:2–5. Birss, V. I., and G. A.Wright. 1981. The potentiodynamic formation and reduction of a silver sulfide monolayer on a silver electrode in aqueous sulfide solutions. Electrochimica Acta27:1–7. Buchwald, V. F.1977. The mineralogy of iron meteorites. Philosophical Transactions of the Royal Society (London) A286:453–91. Buchwald, V. F., and R. S.Clarke, Jr.1989. Corrosion of Fe-Ni alloys by Cl-containing akageneite (beta-FeOOH): The Antarctic meteorite case. American Mineralogist74:656–67. Campbell, G. D., F. J.Lincoln, G. P.Power, and I. M.Ritchie. 1982. The anodic oxidation of silver in sulfide solutions. Australian Journal of Chemistry35:1079–85. Daubr�e, G. A.1862. Contemporary formation of copper pyrite by the action of hot springs at Bagnes-de-Bigorre. Bulletin Societ� G�ologique. 19:529. Djurle, S.1958. An x-ray study of the system Ag-Cu-S. Acta Chemica Scandinavica12:1427–36. Duncan, S. J., and H.Ganiaris. 1987. Some sulphide corrosion products on copper alloys and lead alloys from London waterfront sites. In Recent advances in the conservation and analysis of artifacts, ed.J.Black. London: Summer Schools Press, University of London. 109–18. Fabrizi, M., H.Ganiaris, S.Tarling, and D. A.Scott. 1989. The occurrence of sampleite, a complex copper phosphate, as a corrosion product on copper alloy objects from Memphis, Egypt. Studies in Conservation34:45–51. Fisher, W. W., and A. C., Lasaga. 1981. Irreversible thermodynamics in petrology. In Reviews in Mineralogy. Vol. 8, Kinetics of geochemical processes. Washington, D.C.: Mineralogical Society of America. Garrels, R. M., and J. C.Christ. 1965. Solutions, minerals, and equilibria. San Francisco: Freeman, Cooper. Geilmann, W.1956. Leaching of bronzes in sand deposits (in German). Angewandte Chemie68:201–11. Gettens, R. J.1963a. The corrosion products of metal antiquities. In Annual report to the trustees of the Smithsonian Institution for 1963. 547–68. Gettens, R. J.1963b. Addendum to Mineral alteration products on ancient metal objects (with supplement). International Council of Museums Committee for Scientific Museum Laboratories. Moscow. 63/29. Gettens, R. J.1969. The Freer Chinese bronzes. Vol. 2, Technical studies. Washington D.C.: Smithsonian Institution, Freer Gallery of Art. Goldschmidt, J.1953. A simplexity principle. Journal of Geology61:539–51. deGouvernain, M.1875. Sulfiding of copper and iron by a prolonged stay in the thermal spring at Bourbon l'Archambault. Comptes Rendus80:1297–1300. Hack, H., H.Shih, and H. W.Pickering. 1986. Role of the corrosion product film in the corrosion protection of Cu-Ni alloys in seawater. In Surfaces, inhibition, and passivation., ed.E.McCafferty and R. J.Brodd. Pennington, N.J.: Electrochemical Society. 355–67. Heimann, R. B.1989. Assessing the technology of ancient pottery: The use of ceramic phase diagrams. Archeomaterials31:123–48. Kato, C., and H. W.Pickering. 1984. A rotating disk study of the corrosion behavior of Cu-9�4Ni-1�7Fe alloy in air-saturated aqueous NaCl solution. Journal of the Electrochemical Society131:1219–24.
Kato, C., H. W.Pickering, and J. E.Castle. 1984. Effect of sulfide on corrosion of Cu-9�4Ni-1�7Fe alloy in aqueous NaCl solution. Journal of the Electrochemical Society131:225–12. Kracek, F. C.1946. Phase relations in the system silver-sulfur and the transitions in silver sulfide. Transactions of the American Geophysical Union27:367–74. Little, B.J., P. A.Wagner, W. G.Characklis, and W.Lee. 1990. Microbial corrosion. In Biofilms, ed.W. G.Characklis and K. C.Marshall. New York: John Wiley and Sons. 635–70. Lucey, V. F.1967. Mechanism of pitting corrosion of copper in supply waters, British Corrosion Journal2:175–85. McNeil, M. B., J.Jones, and B. J.Little. 1991. Mineralogical fingerprints for corrosion processes induced by sulfate-reducing bacteria. NACE Paper no. 580. National Association of Corrosion Engineers. Miller, J.D.A., and A. K.Tiller. 1970. Microbial corrosion of buried and immersed metal. In Microbial aspects of metallurgy, ed.J.D.A.Miller. New York: American Elsevier. 61–106. Mohr, D. W. and M. B.McNeil. 1992. Use of modified log-activity diagrams to elucidate type 1 pitting of copper within chloride-bearing waters. Journal of Nuclear Materials190:329–42. Mond, L., and G.Cuboni. 1893. On the nature of antique bronze patina (in Italian). Atti de la reale Accademia dei Lincei, 5th ser.2:498–99. Mor, E. D., and A. M.Beccaria. 1975. Behaviour of copper in artificial seawater containing sulphides. British Corrosion Journal10:33–38. North, N. A. and I. D., MacLeod. 1986. Corrosion of metals. In Conservation of archaeological objects, ed.C.Pearson. London: Butterworths. 69–98. North, R. F., and M. J.Pryor. 1970. The influence of corrosion product structure on the corrosion rate of Cu-Ni alloys. Corrosion Science10:297–311. Pollard, A. M., R. G.Thomas, and P. A.Williams. 1989. Synthesis and stabilities of basic copper(II) chlorides atacamite, paratacamite, and botallackite. Mineralogical Magazine53:557–63. Pope, D. H.1985. MIC in U.S. industries: Detection and prevention. In Proceedings of the Argentine U.S. Workshop on Biodeterioration. Sao Paulo, Brazil: Aquatec Quimica. (CONICET-NSF) 105–18. Pope, D. H.1986. A study of microbiologically influenced corrosion in nuclear power plants and a practical guide for countermeasures. Palo Alto, Calif.: Electric Power Research Institute. Postgate, J. R.1979. The sulphate-reducing bacteria. Cambridge: Cambridge University Press. Pourbaix, M.1966. Atlas of electrochemical equilibria in aqueous solutions. Houston: National Association of Corrosion Engineers. Reiber, S. H.1989. Copper plumbing surfaces. Journal of the American Waterworks Association81:114–22. Roy, R., A. J.Majumdar, and C. W.Hulbe. 1959. The Ag2S and Ag2Se transitions as geologic thermometers. Economic Geology54:1278–80. Sato, S.1982. Case studies in pitting corrosion failures of copper tubes in hot water. Corrosion Engineering (Japan) 31:3–11. Scott, D. A.1985. Periodic corrosion phenomena in bronze antiquities. Studies in Conservation30:49–57. Scott, D. A.1990a. Bronze disease: A review of some chemical problems and the role of relative humidity. Journal of the American Institute for Conservation29:193–206. Scott, D. A.1990b. Private communication. Getty Conservation Institute. Shcherbina, V. V.1978. The geochemistry of monovalent sulfides (in Russian). Geokhimiya10:1444–51. Sinclair, J. D.1982. The tarnishing of silver by organic sulfur vapors. Journal of the Electrochemical Society129:33–40.
Swartzendruber, L. J., L. H.Bennett, and M. B.Mc-Neil. 1973. On the electron configuration theory of marine corrosion. In Proceedings of the Third International Conference on Marine Corrosion and Fouling. Washington D.C.: National Bureau of Standards. 410–26 Syrett, B. C.1977. Accelerated corrosion of copper in flowing pure water contaminated with oxygen and sulfide. Corrosion33:257–62. Syrett, B. C.1981. The mechanism of accelerated corrosion of copper-nickel alloys in sulphide-polluted seawater. Corrosion Science21:187–209. Volpe, L., and P.J.Peterson. 1989. The atmospheric sulfidation of copper in a tubular corrosion reactor. Corrosion Science29:1179–86. Wagman, D. D., W. H.Evans, V. B.Parker, R. H.Schumm, I.Halow, S. M.Bailey, K. L.Chumey, and R. L.Nuttall. 1982. The NBS tables of chemical thermodynamic properties: Selected values for inorganic and C1 and C2 organic substances in SI units. Journal of Physical and Chemical Reference Data 11, supplement #2. Walker, G. D.1977. An SEM and microanalytical study of in-service dezincification of brass. Corrosion33:252–56. Woods, T. L. and R. M.Garrels. 1966. Phase relations of some copper hydroxy minerals. Economic Geology81:1989–2007. Zen, E.-A.1966. Construction and pressure-temperature diagrams for multicomponent systems after the method of Schreinemakers geometric approach. U.S. Geological Survey Bulletin 1225. Zolotarev, E. I., A. P.Pchel'mikov, Ya. B.Skuratnick, M. A.Dembrovskii, N. I.Khokhlov, and V. V.Losev. 1987. Kinetics of dissolution of copper-nickel alloys, anodic dissolution of Cu-30%Ni under steady state conditions (in Russian). Zashchita Metallov23:922–29. AUTHOR INFORMATIONMICHAEL B. McNEIL, B.A., M.A., Ph.D., is presently a metallurgist, Office of Research, U.S. Nuclear Regulatory Commission. From 1987 to 1991 he was at the Naval Coastal Systems Center, Panama City, Fla. His principal research interest is the interpretation of mineralogical and morphological observations to obtain mechanistic information on long-term corrosion processes, especially those arising from biological processes. Address: Office of Research, U.S. Nuclear Regulatory Commission, Washington, D. C. 20555. BRENDA J. LITTLE, B. S., Ph.D., is a research chemist for the Naval Research Laboratory. Her research is directed toward the investigation of microbiologically influenced corrosion of metals and alloys in marine environments. She is the author of numerous journal articles, technical papers, and book chapters on the subject. Address: Naval Research Laboratory, U. S. Navy, Stennis Space Center, Miss. 39529-5004.
 Section Index Section Index |