FOURIER TRANSFORM INFRARED SPECTRAL ANALYSIS OF NATURAL RESINS USED IN FURNITURE FINISHESMichele Derrick
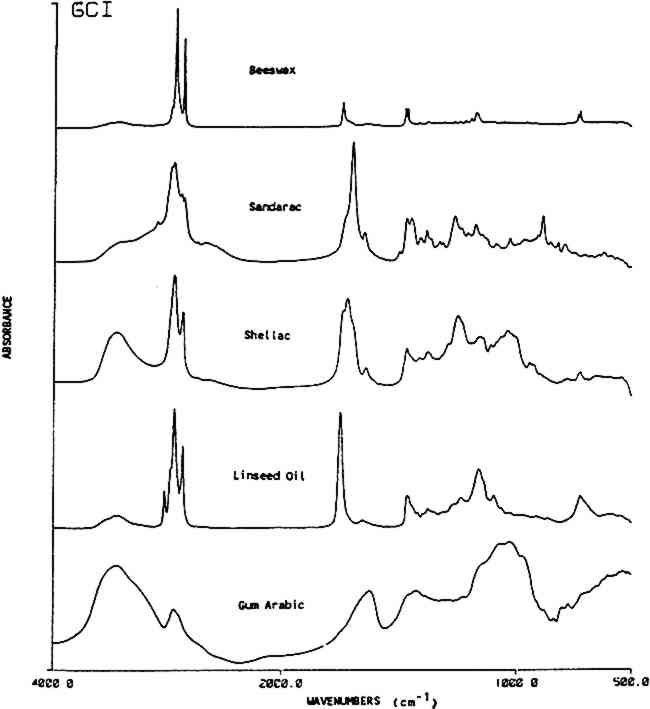
ABSTRACT—Infrared spectroscopy is a valuable method for the detection and identification of organic coating materials. The vibrational bands that appear in the infrared spectra provide information on chemical functional groups of a sample which may allow for general characterization of the material or even the identification of specific compounds. Mixtures of natural materials, as are often found in furniture finishes, can stretch conventional infrared spectroscopy to its limits as an analytical method. This paper outlines the capabilities and limitations of Fourier transform infrared spectroscopy for the identification of natural resins used in historic furniture finishes. 1 INTRODUCTIONFOURIER TRANSFORM INFRARED (FT-IR) SPECTROSCOPY requires minimal sample amounts to provide reliable characterization of the major components in a mixture. With the aid of computer techniques such as spectral subtraction (Koenig, 1975) and deconvolution (Kaupinnen et.al., 1981), the intrinsic information from the spectra can be used to identify even minor components of a mixture. FT-IR has significant advantages over less accurate dispersive spectrophotometers (Low and Baer, 1977). The Fourier transform instrument produces a high resolution spectrum in a very short scan time. This rapid scan time allows for the integration of a large number of scans, thus effectively averaging any noise and yielding spectra with a very high signal-to-noise ratio. Accuracy and reproducibility of spectral band position is achieved in the FT-IR instrument by internal laser calibration (Koenig and Tabb, 1974). The usual procedure for identifying an unknown by its infrared spectrum is to visually match band positions and their relative intensities with the spectrum of a known material. This can easily be done for a pure sample using a computer search program to rapidly compare an unknown to a digitized library of several thousand known pure compounds. However, because unknown samples are often impure, erroneous spectral matches can result. A spectrum for a mixture of materials contains the vibrational bands for each component, therefore a method for separating the bands must be used to identify the individual compounds. This can be done either by separating the mixture components prior to analysis, such as with solvent extraction (Mills, 1972), or by carefully sorting out the bands in the complicated spectra of the mixture. In the latter approach, the major infrared absorption bands are first used to identify the general class or classes of the components. Then, specific band positions of the unknown are compared with bands from reference materials (Smith, 1979). This investigation used the method of spectral identification of compounds in a mixture by band position to study natural resins typically used in historic furniture finishes. Brachert (1978) gave detailed recipes for historical furniture finishes. The five most often used resins (shellac, sandarac, mastic, copal and rosin) were selected from Brachert's compilation of recipes (see Table 2 for frequency of use). A variety of infrared analyses was done on these five resins. First, sets of known resins were used to check the band position variability due to sample preparation, sample size, resin supplier, varnish preparation (solvent extraction and filtration) and resin aging Table 2 Identification Key For Infrared Spectra Of Natural Resins Absorption bands represented as wavenumbers (� 2.6 cm−) 2 EXPERIMENTAL2.1 InstrumentationSPECTRAL ANALYSES WERE MADE on a Digilab 15-E Fourier Transform Infrared (FT-IR) spectrometer equipped with a Motorola 3200 computer. The spectrometer was purged with dry clean, CO2-free air from a Balston 75-60 filtration unit. A Mercury-Cadmium-Telluride (MCT) detector, cryogenically cooled, was used to examine the region from 4000-500 cm−1 (2.5–20 μm). Each spectrum is the sum of two hundred scans collected at a resolution of 4 cm−1. 2.2 SamplesReference resins were obtained from A.F. Suter & Co and the conservation laboratories at the J. Paul Getty Museum. Homogeneous samples were obtained by crushing and thoroughly mixing a 2–3 gram portion, which was then stored in a glass vial prior to analysis. Naturally aged resin samples were obtained from the Harvard Art Museums' collection of Gettens and Stout materials. The resins were films (<20 μm thick) on glass plates. The dates on these plates were from 1933–1937. It is assumed that the plates, which were labeled as controls, were stored in a drawer during the last 5 decades. A 50 mm2 section of each resin was removed from the plate with a scalpel and placed in a clean glass vial with a Teflon™-lined cap. The unknown sample set was composed of finishes on furniture pieces from the J. Paul Getty Museum. Each piece was of 18th century European manufacture and was typically of veneer on oak. Samples were arbitrarily chosen from obscure areas on the furniture, near or under mounts, in the rear or on the legs. A scalpel was used to scrape barely visible amounts of sample (<100 μg) from clean, surface layers of the finish. This minimized the chance of contamination from dirt and wood particles which can interfere with the interpretation of the resin spectra. In a multilayered finish, this sampling method selectively removes the more recent coatings. In these cases, more than one sample may be scraped from different depths. 2.3 Sample PreparationSamples were prepared as KBr pellets in either the standard 13 mm size or 1.5 mm micropellet size. Approximately 1 mg of a crushed bulk resin sample was used for the 13 mm KBr pellets. These standard sized pellets were used in the tests on variability due to sample preparation, resin supplier and resin refinement. Additionally, the 13 mm pellets were used for the resin mixtures; each mixture had a total weight of 1 mg with the individual components weighing from 0.01 mg to 1.20 mg. The 1.5 mm micropellets were prepared from approximately 30 μg of sample in a glove bag under a dry nitrogen purge. The micropellets were mounted in a Harrick Scientific 4� beam condenser for infrared analysis. The natural and artificially aged samples, the solvent dependency study and the furniture finish samples were all analyzed by this micropellet technique. Because the sample sizes were sufficient and the samples were prepared in the purged glove bag, none of the possible problems with KBr pellets (external contamination, water absorption, sample reactions) applied to this study. For the solvent dependency study, 5 wt/v% solutions were prepared with sandarac, mastic and rosin in ethanol, turpentine and mineral spirit solvents and with shellac and copal in ethanol. After 48 hours, the solutions were filtered through one layer of cheesecloth, then 20–30 ml of each solution was cast onto clean thin sheets of aluminum using 6“�6” Teflon™ molds (Hansen and Taketomo, 1988). The films were air-dried for approximately one week, then analyzed. For the accelerated deterioration studies, a thin layer of four resins in ethanol (mastic-20 wt/v%, rosin-20 wt/v%, sandarac-33 wt/v% and shellac-33 wt/v%) were painted with an ethanol-cleaned paint brush onto clean, polished-surface aluminum plates. These samples were allowed to dry overnight in a fume hood, then placed in a Weather-Ometer with controlled light, heat and humidity conditions. The samples were irradiated with a xenon lamp set at 0.35 W/m2 (340 nm) with inner and outer borosilicate filters simulating outdoor sunlight (Brennan, 1987). The black panel temperature was 53�C and the relative humidity was 50%. The total exposure for the samples ranged from 0 to 151 kJ (151 kJ is equivalent to 120 hours exposure at an irradiance of 0.35 W/m2 at 340 nm). The resin controls were kept in the dark for an equivalent time period. 2.4 Computer MethodsFor each sample, the computer printed out band position values for peak maxima. In regions where peaks are overlapped, a deconvolution program was used to elucidate the individual features. The program operates on two or more overlapping bands to reduce the line width of the individual components and therefore improve the resolution of each line. Spectral subtraction methods were used to separate components of a mixture. Intense bands were used to identify major components whose pure spectra were then multiplied by a scaling factor and subtracted out. After spectrally removing the bands corresponding to the major component, a scale expansion was used to enhance the remaining bands and identify the minor components. Following the same general scheme, computer addition of individual spectra was used to simulate spectra of mixtures, such as varnish recipes, which were then stored in a search library for access by the computer. 3 RESULTS AND DISCUSSION3.1 General ClassificationINFRARED SPECTROSCOPY PRODUCES a characteristic spectrum for any compound dependent on the functional groups that are present. For identification purposes, natural products may be grouped into the following classes that contain similar IR active functional groups: resins (tree), resins (insect), oils, gums and waxes. Table 1 shows these together with specific peak positions that occur within each material class. Figure 1 gives exemplary spectra of each category. The spectra of each of these classes are readily identifiable by their characteristic functional group vibrations. Typically OH stretches occur as broad bands near 3400 cm−1, methyl and methylene stretches occur in the region of 2800–3200 cm−1 and carbonyl bands are seen at 1690–1750 cm−1. The bands in the fingerprint region (below 1800 cm−1) are due to C-H, C-O and C-C wags and bends. The exact location (wavenumber) for each band depends on its molecular environment. More information on vibrational group assignments for these natural materials may be found elsewhere (Bellamy, 1968 and Omecinsky and Carriveau, 1986).
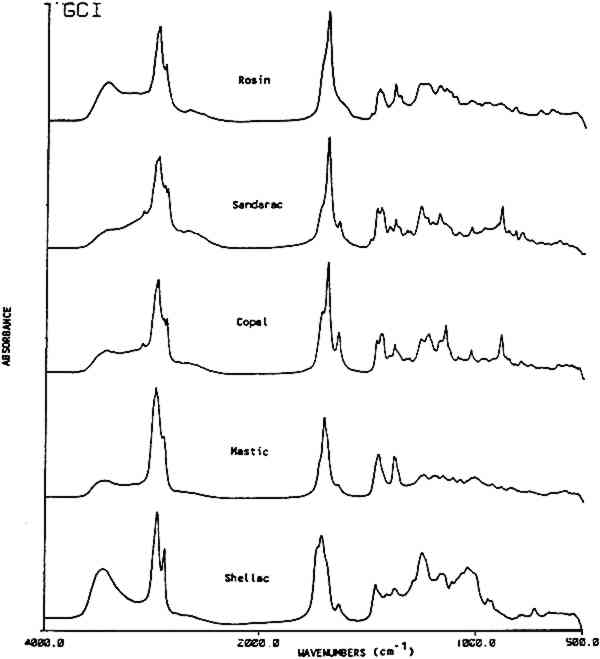
Table 1 This table lists characteristic absorption bands for several classes of natural materials. These bands appear consistently in the spectra of each class of material along with other bands, not in this list, which are specific to the individual types of materials found within each class. 3.2 Resin Identification KeyTable 2 gives an IR absorbance band identification key for five commonly used natural resins in furniture finishes along with their family name and major components. Additional information is included on the use of these resins in historic finishes. Figure 2 shows an infrared absorbance spectrum for each resin examined. Once a natural material has been classified as a resin using the information in Table 1, then the type of resin may be distinguished using the additional information provided in Table 2.
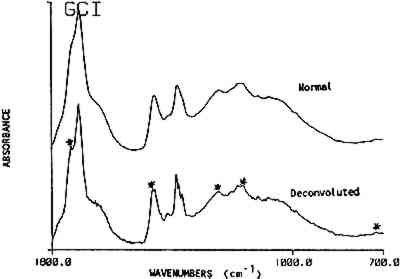
This key provides a simplified structure for the easy identification of the presence of a resin in a spectrum by a list of its absorbance band positions. The strongest bands in the resin spectra are generally the carbonyl and hydrocarbon stretching frequencies. These bands, listed at the top of the key can provide a distinction for four of the resins. Sandarac and copal are chemically and spectrally very similar and may 3.3 ReproducibilityThe premise of the identification of resins by band position alone is based on the assumption that the frequency of the absorption bands is reproducible. Several factors were examined individually to assess band position variability. They were: 1) instrument stability; 2) sample preparation methods; 3) source or degree of refinement of the resin; 4) sample size; 5) preparation of a varnish; and 6) the extent of deterioration or oxidation of the resin. A summary of standard deviations for the band position Table 3 Band Position Precision For Several Parameters Instrument stability was determined using the reference polystyrene film which had a precision of 0.1 cm−1 over a period of three years. Spectra were generated from a standard film using direct transmission which required no sample preparation. Reproducibility in sample preparation was then checked by preparing several pellets from the same batch of mastic resin which gave variations of 1.1 cm−1. Additionally, variations in wavenumber due to sample size were examined. Sandarac bands, using sample sizes ranging from 8 μg to 1.8 mg, were found to vary only 0.3 cm−1. This higher precision may be attributed to the sharper bands in the sandarac spectra or to the possibility that the sandarac bulk sample was more homogeneous than mastic. Differences due to the resin's source or supplier were checked by analyzing 8 mastic samples obtained from different suppliers. Additionally, 14 samples of shellac were analyzed, not only from different suppliers, but also with varying degrees of processing or refinement ranging from seedlac to dewaxed, decolored shellac. The band position reproducibility was 1.2 cm−1 for both sets of samples. Varnish preparation involved dissolution of the resins in a solvent, then filtration or sedimentation to remove insoluble fractions. Cast films of the resins prepared from different solvents gave an average variability of 0.8 cm−1. While some residual solvent was seen in the spectra of the films cast from turpentine, the spectra of the resins were still easily recognizable by their band positions. In general, spectra of the resins after solvation and filtration contained slightly sharper and better resolved bands than those prepared from the ground resins. Since finishes found on pieces of furniture in a museum may be of considerable age, the stability of the band positions in the IR spectrum of a resin as it deteriorates was considered. Naturally aged resin films obtained from the Gettens and Stout collection were analyzed, in addition to new resin films for which the photochemical deterioration process was accelerated by exposure to a xenon lamp. Overall, the naturally aged resins showed an average deviation of 1.3 cm−1, while the xenon light exposed resins showed a deviation of 2.1 cm−1. A comparison of the spectra of the naturally aged samples versus light and heat exposed samples will be covered in another paper (Derrick and Hansen, 1988). The most important factor for this paper is that the band positions are stable and may still be used to identify each of the resins after they have been degraded. 3.4 Resin MixturesUp to this point, reproducibility of the band positions has been demonstrated as a function of several variables. The resin identification method was then applied to resins in mixtures. Combinations of two or more resins are given in 80 recipes in Brachert's paper. In this study, resin mixtures were prepared in the proportions specified by six recipes. In addition, 44 mixtures of sandarac, copal, mastic, rosin After the infrared analysis of each mixture, a list of band positions was generated. The spectrum was then deconvoluted and a new band position list was generated. Only the deconvolution band positions were used in this resin mixture section. It was found that when a resin accounts for at least 50% of the total mixture a deconvolution step was unnecessary, i.e. all the bands required for the identification of the resin were present whether or not the spectrum had been deconvoluted. However, as the concentration of a resin decreased within the mixture, the deconvoluted spectrum consistently provided a higher proportion of the peaks necessary for an accurate identification of the resin. The 5 resins examined in the mixtures could be detected at concentrations as low as 2 w/w%. At these low levels, approximately half of the bands for a resin were present in the deconvoluted spectrum of the mixture. At resin concentrations above 6 w/w%, most (>90%) of its bands could be found in the deconvoluted spectra. Figure 3 shows a spectrum of mastic/oil film (Gettens and Stout collection, dated 1934, 5cc oil in 100cc ethanol with 33g mastic) before and after deconvolution. The bands specific for oil, visible only after deconvolution, are marked.
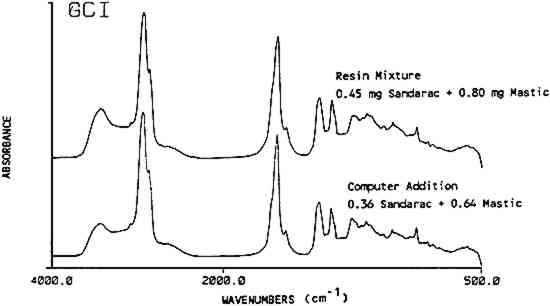
Copal was the one exception to the identification of a resin in a mixture by its band positions. The peaks used for the identification of copal and sandarac overlap Using mixtures of up to four components, it was apparent that the maximum number of resins that can be identified in one finish is probably four. More complex mixtures should be examined by another method, such as chromatography. 3.5 Spectral Subtraction and AdditionComputer assisted spectral subtraction techniques were employed to remove the major peaks in a mixture's spectrum revealing the presence of trace components. For one mixture containing 99.2 w/w% sandarac and 0.8 w/w% mastic, no mastic peaks were found in the deconvoluted spectra. Spectral subtraction of a known spectrum of pure sandarac was applied to the original spectrum, not on the deconvoluted spectrum, to remove 90% of the intensity of the sandarac-related bands. Afterwards the subtracted spectra were deconvoluted and examined for the presence of mastic bands. Half of the expected mastic bands were found confirming that mastic was indeed present. One problem can result in the subtraction spectra. Variations in line widths between the two spectra on which the subtraction is performed can result in negative baseline aberrations. In this case, a peak due to mastic may still not be present in the subtracted spectrum if it was close to a large sandarac peak. Alternatively, the peaks that do appear may be shifted outside the normally expected band range. This problem may be lessened by not subtracting the total amount of the major component. Actually, it only appears necessary to subtract enough of the major component to bring the minor component up to the equivalent of 10–20% composition. Another use for the computer is to synthesize spectra of resin mixtures via spectral addition methods. This procedure could be used to efficiently create a large reference set of spectra for typical furniture finish mixtures. This method was tested using a computer synthesized mixture of 64% of a mastic spectrum added to 36% of a sandarac spectrum in order to emulate a previously prepared mixture of 0.80 mg mastic and 0.45 mg sandarac. The spectrum for the actual mixture and the computer synthesized mixture corresponded well (Figure 4). A computer search using the Sadtler Library programs was made of the synthesized spectra versus this laboratory's library files consisting of over 300 natural and synthetic materials including all resin mixtures used in this study. The best match for the search was the actual resin mixture which the synthesized spectra were trying to emulate. The corresponding hit index was 0.0716 on a scale where 0.0000 is a perfect match and a typical match of 0.1500 is considered a good fit. In addition, mastic was a component in each of the ten best hits and sandarac was present in six of these. The search programs take the difference of the spectra in question versus the reference spectra and match according to the least differences. Computer synthesized spectra appear to be a viable, time saving alternative to manual preparation of resin mixtures.
3.6 Unknown samplesInfrared analysis was then done on samples taken from 18th century European furniture, even though it was unlikely that the outer layers sampled from the finishes contained the original resins. Samples from the exterior of two tables (72.DA.49, 84.DA.77), a cabinet (77.DA.91), a cupboard (72.DA.39), a commode (55.DA.5) and a desk (67.DA.10) were found to be shellac, while the finishes on a rolltop desk (72.DA.47), a double desk (70.DA.87) and a music stand (85.DA.216) were found to be mixtures of shellac and cellulose nitrate. Additional bands corresponding to oil were found on the rolltop desk, and another sample taken from the inside of a drawer on this desk was found to contain only shellac. For each sample the major components were identified, then the spectra were deconvoluted to check for minor components. This was followed by subtraction of the major component to further examine the spectra for minor components. According to Mattiello (1941), cellulose nitrate was often added to shellac in the 1930's and 40's to improve its water resistance. However, from these samples, it 4 CONCLUSIONSFOURIER TRANSFORM INFRARED ANALYSIS can identify five natural resins (shellac, sandarac, mastic, copal and rosin) individually and as components in a mixture based on a set of band positions presented in this paper. The precision of the band positions was demonstrated to be within �2.6 cm−1 for: 1) the instrument; 2) sample preparation used in this study; 3) variations due to resin supplier or degree of refinement. Changes in band position due to varnish preparation and photochemical aging were shown not to diminish the effectiveness of the band identification scheme. For resin mixtures of two components, with a total sample mass of approximately 1 mg, a component with a concentration of 50 w/w% or greater may be identified by the presence of the band positions listed in Table 2. Minor components in a resin mixture may be identified by computerized deconvolution of the spectra to resolve any overlapping bands. With deconvolution, a component with a concentration as low as 6 w/w% may be readily recognized and components with concentrations as low as 2 w/w% may be tentatively recognized. Additionally, the computer may be used to spectrally subtract most of the major component from a mixture, thus, in effect increasing the relative proportion of the minor components. Using computer subtraction followed by deconvolution, components in concentrations as low as 1 w/w% may be identified. ACKNOWLEDGEMENTSI WOULD LIKE TO THANK Barbara Roberts and Gillian Wilson from the Getty Museum for allowing samples to be taken from museum objects and for Barbara's help and encouragement. I would also like to thank Marjorie Cohn and Eugene Farrell for their permission for, and Johann Peron for her help in, sampling the Gettens and Stout collection at the Harvard Art museums. I appreciate Robert Mussey for generously sharing his translation of Brachert's article. I would like to thank Eric Hansen for his advice on the accelerated aging of resin films. I am grateful to Frank Preusser, Neville Agnew, Jack Gromek, John Perkins, George Rogers, David Scott and Dusan Stulik for their very helpful comments and contributions. BIBLIOGRAPHYBeck, C.W, Wilbur, E., Meret, S., Kossove, D. and KermaniK.“The Infrared Analysis of Amber and the Identification of Baltic Amber.”Archaeometry, 9, 1965, pp. 96–109.
Bellamy, L.J.Advances in Infrared Group Frequencies, New York: Barnes and Noble, 1968. Brachert, T.“Historical Transparent Varnishes and Furniture Polishes.”Maltecnik-Restauro, no. 1–5, 1978. translated by R. Mussey. Brennan, P. and Fedo, C.“Sunlight UV and Accelerated Weathering,” presented at the Society of Plastics Engineers Automotive RETEC, Nov. 1987. Derrick, M., and Hansen, E.“Comparison by Infrared Spectroscopy of Natural and Accelerated Aging of Natural Resins,” in preparation. Gianno, R., Von Endt, D., Erhardt, W., Hopwood, W., and Kochummen, K.“The Identification of Insular Southeast Asian Resins and other Plant Exudates for Archaeological and Ethnological Application.” Jubilee Conservation Conference Papers, Univ. of London, Institute of Archaeology, 1987. Hansen, E. and Taketomo, A.“Technical Note on the Preparation of Free Standing Films.” submitted to Studies in Conservation. Kauppinen, J., Moffatt, D., Mantsch, H. and Cameron, D.“Fourier Self-Deconvolution: a Method for Resolving Intrinsically Overlapped Bands.”Applied Spectroscopy, 35(3), 1981, pp. 271–276. Koenig, J.L. and Tabb, D.L.“Application of Fourier Transforms Infrared Spectroscopy”, Canadian Research and Dev., (Sept.-Oct. 1974). Koenig, J.L.“Application of FTIR Spectroscopy to Chemical Systems.”Applied Spectroscopy, 29(9), 1975, pp. 193–309. Low, M.J.D. and Baer, N.S.“Application of Infrared Fourier Transform Spectroscopy to Problems in Conservation: General Principles.”Studies in Conservation, 22, 1977, pp. 116–128. Mattiello, J.“Protective and Decorative Coatings: Paints, Varnishes, Lacquers and Inks.” Vol. 1, Raw Materials for Varnishes and Vehicles, New York: Wiley and Sons, Inc., 1941. Mills, J.S.“Identification of Organic Materials in Museum Objects.”Conservation_in the Tropics, O.PAgrawal (ed.), 1972, pp. 159–170. Mills, J.S. and White, R.“Natural Resins of Art and Archaeology: their Sources, Chemistry and Identification.”Studies in Conservation, 22, 1977, pp. 12–31. Omecinsky, D., and Carriveau, G.W.“IR Investigation of Resinous and Synthetic Varnishes.” AIC 10th Annual Meeting Preprints, Milwaukee, 1982, pp. 141–149. Shearer, J.C., Peters, D.C., Hoepfner, G. and Newton, T.“FTIR in Art Conservation.”Analytical Chemistry, 55, 1983, pp. 874A–879A.
 Section Index Section Index |