Polymer, 1983, Vol. 24 June 1983. pp. 651-666
(Received 17 November 1981)
The structure and properties of gelatin in the solid state are discussed with respect to the structural, chemical and polymeric characterization of gelatin. Also the influence of casting conditions on the structural characteristics of gelatin, the relationship between the physico-mechanical properties of gelatin and the conformational state of the gelatin macromolecules and the effect of water on the structural and mechanical properties of gelatin are discussed. The effect of heat on the structural transformations and properties of gelatin are considered with reference to structural transformations, physico-mechanical properties, physico-chemical properties, internal stress and relaxation properties. The principles of modification of the physico-mechanical properties of gelatin are discussed, in particular the physical modification, physico-chemical modification and finally chemical modification.
Keywords Polymer; solid; gelatin; structure; properties; modification
It is well known that gelatin is a product of the structural and chemical degradation of collagen. Because of this, reviews and monographs dealing with the structure and properties collagen1-6 also present data on gelatin thus providing additional information on collagen. Moreover, the few available reviews and monographs devoted specifically to gelatin7-9 deal, to a large extent, with the structure and properties of collagen. This naturally stems from the similarities in amino acid composition and, to some extent, in structure between collagen and gelatin and hence the similarity in properties between the two proteins At the same time, gelatin exhibits its own properties which are either absent in collagen or are developed only to a slight extent. Gelatin has some individual importance as a polymeric product widely utilized in the manufacture of various articles and materials10-18. In most cases, except for the food industry, gelatin is used in the solid state. However, whereas the properties of gelatin gels and solutions have been thoroughly surveyed19-26, only a few reviews have dealt with the solid state of gelatin9,27-29 and they provided only a brief outline, ignoring many aspects of the behaviour and properties of gelatin.
Alongside a large number of quite useful features inherent in gelatin, it also exhibits some significant shortcomings which are aggravated under unfavourable temperature and humidity conditions. Among these shortcomings there is, first of all, a high brittleness exhibited by solid gelatin at reduced humidity and elevated temperatures, the brittleness resulting in premature failure of gelatin articles. Because of this, the problem of modification of the properties of solid gelatin and, first of all, the problem of its plasticization has been the subject of much investigation for over a century and cannot yet be considered as completely resolved. This impelled us to discuss the methods for modifying the properties of solid gelatin and to compare these methods and outline their capabilities in reducing brittleness of gelatin materials under severe environmental conditions.
The characterization of gelatin as a product of the denaturation and structural degradation of collagen has been discussed not only in numerous experimental works but also in some review papers and monographs4,7,27,30-32. The physico-chemical and structural characterizations of gelatin have been given in refs. 7, 26 and 27, and the polymeric characterization in the fundamental review by Yannas9 and the paper by Kargin et al.20.
The properties of gelatin as a typical rigid-chain high molecular weight compound are in many respects similar to those of rigid-chain synthetic polymers. Gelatin exhibits essentially the same common properties typical of polymeric substances, which is not the case with native collagen. Thus, in a similar way to linear-chain synthetic polymers, in aqueous solutions gelatin macromolecules assume, at elevated temperatures, the conformation of a statistical coil7. Under specific conditions (temperature, solvent, pH) gelatin macromolecules can display a flexibility sufficient to realize a wide variety of conformations. This makes it possible to vary all the gelatin characteristics dependent on its molecular
structure. Besides, gelatin, similar to synthetic high polymers, shows a rather wide molecular weight distribution7,33. Quite as interesting is the capacity of gelatin to form a large variety of supermolecular structures, from the simplest globular structure 34, typical of amorphous polymers35, to a well developed fibrillar structure with various intermediate states36-43. Clearly the differences in supermolecular structures should be reflected in the physico-mechanical properties of the gelatin materials.
Structural diversity of gelatin chain units determines the specific features of gelatin properties. Most synthetic polymers show no such features that are typical of most biopolymers.
The first peculiarity of gelatin common to all biopolymers arises from the presence of both acidic and basic functional groups in the gelatin macromolecules.
The second peculiarity of gelatin is its capacity to form a specific triple-stranded helical structure not observed in synthetic polymers (this structure is formed in solutions at low temperatures). The rate of the formation of a helical structure depends on many factors such as the presence of covalent cross-bonds7,43, gelatin molecular weight44, the presence of iminoacids45 and the gelatin concentration in the solution 46,47.
The third peculiarity of gelatin as a biopolymer is its specific interaction with water which is different to that observed with synthetic hydrophilic polymers. This peculiarity governs the structural and physico-mechanical properties of gelatin in the solid state and will be discussed in a separate section of this paper.
The details of the casting process are known to have a pronounced influence on the polymer structure in the finished products. This is especially true of the manufacture of gelatin films or layers. Thus the conformational state assumed by gelatin, macromolecules is completely determined by the temperature of casting, the rate of drying, the concentration of gelatin in the initial solution, as well as by the nature of the solvent and the content of various denaturating or structuralizing substances.. X-ray diffraction48-52, i.r. spectroscopic53 and optical rotation50,53-55 studies of gelatin films have revealed that in films cast at room temperatures and lower, the gelatin macromolecules have mainly a collagen-like helical structure (hereafter such films will conventionally be referred to as 'cold' films and such gelatin as 'helical' gelatin). At the same time, in films prepared from aqueous solutions by evaporating the solvent off at temperatures above 35°C, gelatin macromolecules assume the conformation of a statistical coil with no indications of ordering (hereafter such films will conventionally be called 'hot' films and such gelatin 'coiled' gelatin)27,53,53,56.
The kinetic nature of the helix formation process25,46 necessitates storage of gelatin gels for a certain period of time prior to drying. During this period, the maximum degree of helicity is attained. The degree of renaturation of the collagen-like helical structure may vary substantially during the casting of the gelatin films and may attain high values in films cast from well aged gels. The degree of helicity of gelatin macromolecules in films is also affected by air humidity52,34,57.
The effect of gelatin concentration in the starting solution on the structure of films is closely related to the temperature conditions of the film formation. Thus, the closer the temperature of drying to the gel melting temperature the higher the gelatin concentration needed to obtain as large a degree of renaturation of the collagen-like helical structure as possible55.
The conformational state of the macromolecules in the solid gelatin (for instance, in films) depends on the presence of some substances in the initial gelatin solution. Thus, the replacement of water by such gelatin solvents as formamide, dimethylsulphoxide (DMSO), ethylenechlorohydrine (ECH) prevents helix formation58,59. The addition of urea or thiocyanates to aqueous gelatin solutions also hinders renaturation of the gelatin. Moreover, treatment of films of helical gelatin with solutions of urea or thiocyanates results in a transition to the coil structure52. At the same time, crosslinking agents may both promote and hinder renaturation of gelatin depending on the relative rates of the helix formation and crosslinking processes60,61.
It thus appears that the structural characteristics of gelatin in the solid state can be controlled at the stage of the gelatin structure formation in solution. This makes it possible to produce gelatin materials with markedly differing structures, i.e. in the conformational states of the constituent macromolecules and hence, as will be shown in the following section, in their physico-chemical and mechanical properties.
The physico-mechanical properties of rigid-chain polymers are known to depend appreciably on their molecular and supermolecular structures. Thus polymers having fibrillar structures display typical polymeric properties, while polymers having globular structures lose these properties partially or completely. This is well exemplified by amorphous polyarylates with globular and fibrillar structures which differ from one another in their physico-mechanical properties drastically62. Naturally, the differences at the molecular (helix-oil) and supramolecular levels should affect the physico-mechanical properties of gelatin materials (Table 1).
* The films underwent brittle fracture
Figure 1 Dependence of the limiting shear stress of gelatin gels on ECH content for various gelation durations: (1) 1 hr; (2) 2 h; (3) 1 day: (4) 4 days
Figure 2 Dependence of resistance to impact (P) of gelatin films on the ECH content in the starting gelatin solutions. P/P0 0 (1); 0.3 (2); 0.65 (3); 0.8 (4) and 0.9 (5)
The mechanical properties of films containing macromolecules of different conformations respond in different ways to changes in relative air humidity. It has been shown51 that coiled gelatin is less strong and less elastic than helical gelatin at 45 to 65% air humidity, though the difference in elasticity decreases as the humidity falls. As air humidity rises (above 65% r.h.) the elasticity of coiled gelatin increases more rapidly than does the elasticity of helical gelatin. The strength of coiled gelatin, however, decreases simultaneously to the extent that the increased elasticity of the films can hardly be utilized, At the same time, films of coiled gelatin cast at 65°C from glycerol solutions exhibit (at 25°C) rubber-like properties: the films may undergo as much as 700% reversible deformations64.
Coiled gelatin films are also formed from aqueous-organic solutions. The mechanical properties of gelatin films obtained from water-formamide mixtures at 20°C are given in Table 265. As can be seen from Table 2, brittleness of gelatin films (their exceedingly low resistance to impact) increases drastically as the degree of helicity decreases. Films prepared from water-DMSO and water-ECH mixtures66 show a similar behaviour, A decrease in the ECH concentration in the starting solution, i.e. a decrease of helicity of gelatin macromolecules58, results in a sharp decrease in gelatin gel strength (Figure 1) and in the corresponding decrease in film resistance to impact at ordinary and reduced moisture contents (Figure 2, curves 1-3). The films prepared from solutions containing more than 10 vol% ECH show an improved resistance to impact only at elevated air humidity (Figure 2, curves 4 and 5). The introduction of DMSO into aqueous gelatin solutions (up to 30% by volume) also results in a dramatic decrease in resistance to impact of films prepared from these solutions66. This was observed with films having an equilibrium water content at <65% r.h. At the same time, strength of gelatin gels, containing DMSO in the percentages indicated above, increases (unlike those with ECH). A decrease in film resistance to impact (at DMSO concentrations below 30%) can be attributed to a decrease of gelatin macromolecules' helicity rather than to destructuralization of the system. Indeed, at the DMSO concentrations indicated, the duration of the induction period of the structure formation process remains constant while the gelatin gel strength increases66. The conformational state of the gelatin macromolecules also influences its physico-chemical properties such as solubility, swelling and sorption capacities67,68.
To sum up, the data on the effect of the conformational state of gelatin macromolecules on the mechanical properties of gelatin products available at present, suggest that a decrease in the degree of helicity of gelatin macromolecules always results in deterioration of the solid gelatin mechanical properties provided gelatin is in the glassy state, i.e. at normal (65% r.h.) and reduced moisture contents.
In the past twenty years the state of water in biopolymers has been discussed extensively in the literature69. As an inherent component of living cells, water plays an important role in all biological processes70-71 Because collagen and gelatin are important commercial products, the elucidation of the effect of water on their structural and mechanical properties is of great practical significance. It is well known that the isotherms of water sorption by gelatin are S-shaped which is typical of most natural and synthetic hydrophilic polymers72-75 and is evidence for the varied degrees of interaction of the polymers with sorbed water76. This S shape has allowed many authors72,75 to apply the Brunauer, Emmett and Teller (BET) polymolecular adsorption theory77 to the gelatin-water system. For this system the sorption isotherm plotted on the BET coordinates exhibits linearity up to a relative vapour pressure of ca. 0.4. A thorough study of the sorption parameters of hydrophilic polymers toward water has shown the linear region to correspond to the strongest interaction between the components of the system76. The amount of water bound directly to gelatin (with the greatest energy effect) is 12-14% and corresponds to the so-called 'monomolecular layer'. Further water sorption ('polymolecular layers') is accompanied by a far lower thermal effect though this water is also bound as will be shown later.
Water sorption by gelatin depends on a number of factors, both common to all substances and specific for gelatin and related polymers. Thus the sorption capacity of gelatin depends substantially on pH and increases with ionization of dissociating groups73,78. At the same time, the sorption capacity of gelatin is independent of its molecular weight73 except probably in the case of a highly hydrolysed product where the number of carboxyl and amino terminal groups is greatly increased, and also the sorption capacity of gelatin decreases with increasing temperature72,79, which is of some practical importance80. The specific behaviour of the gelatin sorption capacity toward water vapours, considered as a function of the conformational state of gelatin macromolecules, is also of interest. Comparison of the sorption capacities of films made of collagen with those of helical and coiled gelatin has shown that the higher the degree of molecular ordering in the protein the higher its sorption capacity toward water vapour at almost all air humidities75. Only at P/P0=0.9 will coiled gelatin become a somewhat stronger water absorbent than its analogues characterized by the higher degrees of molecular ordering. A similar sorption behaviour has been reported in refs. 81 and 82. The analysis of the sorption isotherms in terms of the BET theory and calculation of the amount of water contained in a monolayer, Am, show that biopolymers with the helical structure (both synthetic polypeptides83 and collagen and gelatin75 feature higher Am's than the same substances in a disordered state. The analysis of the literature data on hydration of gelatin and collagen makes it possible to subdivide protein-bound water according to its state, function and influence on the properties of these materials. Hence the critical amount of water corresponding to the optimum physico-mechanical properties of gelatin materials may be determined. Table 3 contains the data reported by various authors on the amount of water bound in helical gelatin and in collagen. As can be seen from Table 3, the amounts of bound water in gelatin and collagen are 0.37 and 0.47g per g of dry protein, respectively. It is precisely at those water contents that the heat of wetting gelatin with water approaches zero103. Protein bound water does not freeze at temperatures down to -60° to -70°C92,97-99 and this water differs from free water in all respects87. Water sorbed by gelatin in quantities above ca. 0.37 g/g and by collagen above ca. O.47 g/g is considered free water possessing properties of bulk water. However, the free water present in collagen is in three states99: the first portion of water, although it freezes, differs from bulk water in its temperature and heat of melting; the next portion differs from bulk water in the melting point only; finally there are portions identical to bulk water in both temperature and heat of melting. This suggests that portions of free water are strongly influenced by protein macromolecules. It is not unlikely that this influence results from intense proton exchange between bound and free water molecules102.
As for water bound by protein it may be subdivided into three types according to its state:
(1) Water bound by high-energy sorption centres, This water occurs inside the collagen triple helix and plays a major role in its stabilization by intramolecular hydrogen bonds. The amount of this water depends on the degree of helicity of the macromolecules and ranges from 0.04 to 0.055 g/g for gelatin95,104 and from 0.10 to 0.12g/g for collagen105,106,
(2) Water sorbed by polar groups of gelatin and collagen macromolecules. This water is also strongly bound with the proteins by H-bonds and is located outside the helical fragments but it also contributes substantially to the stabilization of the collagen helical structure. The amount of this water in gelatin and collagen probably corresponds with the so-called monomolecular layer. The average amount of water necessary to constitute the monolayer calculated from the data of various authors using the BET equation is 0.086g/g for gelatin and 0.105g/g for collagen72,73,75,79,83. Water directly bound with macromolecules of the proteins through H-bonds (both inside and outside helical fragments) should be considered as structural water, which ranges from 0.12 to 0.14 for gelatin and from 0.21 to 0.23 g/g for collagen. The latter value, though obtained by approximate calculations from data of different authors, compares well with the results of calorimetric and X-ray diffraction experiments107,108. This moisture content corresponds to the maximum so-called crystallinity of collagen109-110
(3) Water sorbed by proteins to give polymolecular layers. Proceeding from the total amount of water bound with gelatin and collagen (Table 3) and from the amount of structural water in them, polymolecular layers should account for 0.23-0.25 and 0.24-0.26 g/g of water for gelatin and collagen, respectively.
Table 4 presents the ranges of variation of the water content in gelatin and collagen related to the types of bound water discussed above. It also contains the corresponding relative pressures of water vapour found from the sorption isotherms obtained by various authors. As can be seen from Table 4, the range of structural water extends to P/P0=0.6. According to the i.r. data111 that range corresponds to the maximum ordering of crystalline domains in collagen. Four regions of moisture content (the fourth one corresponding to free water) with the markedly different behaviour of collagen were also found'°'. These regions are fairly close to those presented in Table 4. A similar subdivision of bound water in gelatin was inferred from the step-wise behaviour of the dependence of the long transversal relaxation time (T2a) on the moisture content of gelatin90.
Figure 3 Dependence of resistance to impact (P, kg cm cm3) of cold gelatin films on the pressure of water vapour (1) and the isotherm of water sorption by helical gelatin (2)
A number of authors have thus considered water a necessary element for the formation of the collagen and gelatin helical structures. A definite amount of water is believed to be necessary to maintain the native conformation of collagen molecules4,6,96,99-106,111. The specific role of water in the formation of the helical structure has also been demonstrated for synthetic polypeptides isomorphous to collagen112 The effect of water on the mechanical properties of collagen and gelatin also provides some information, though indirectly, on the role of water in the formation of the collagen and gelatin helical structures. Gelatin dehydrated below a 2% moisture level becomes insoluble in water because of crosslinking between the gelatin macromolecules113. Dehydrated gelatin is thus a crosslinked polymer which makes it extremely brittle and hence unusable in the solid state. At 25±3% moisture content, gelatin undergoes transformation from the glassy to the rubbery state at room temperature114 The behaviour of gelatin in the two states above and below Tc manifests itself most clearly when gelatin is subjected to impact74. Figure 3 shows the dependence of resistance to impact of cold gelatin films on the relative pressure of water vapour (curve 1). The isotherm of sorption of water vapour by helical gelatin is also shown (curve 2). As can be seen from Figure 3, both curves can be subdivided into three regions. In the first region extending up to 0.12-0.13g/g moisture content (region 'a') the resistance to impact of gelatin increases gradually (up to P/P0~0.4) with moisture content. A comparison of this finding with the data in Table 4 shows that the increase results from the uptake of structural water.
In the second region ('b', Figure 3) extending from 0.13 to 0.25 g/g moisture content, resistance to impact of helical gelatin remains practically unchanged (P/P0 ~0.4-0.8). It follows from the analysis of the sorption isotherms in terms of the BET theory and from the data given in Table 4 that when P/P0>0.4 polymolecular layers begin to form. The latter process has no actual effect on intermolecular interactions in gelatin and hence on the mobility of its macromolecules.
Figure 4 Dependence of the resistance to impact (P) of hot gelatin films on the pressure of water vapour (1) and the isotherm of water sorption by coiled gelatin (2)
Figure 5 Dependence of the second moment (1) and long transversal relaxation time (2) on the pressure of water vapour
The third region ('c', moisture content above 0.25 g/g, P/P0 >0.8) shows wide variations in resistance to impact of gelatin films. These variations, however, result from the transitions of the polymer from the glassy state to the rubbery state (an approximate twofold increase of resistance to impact) and then to viscous-flow states (a dramatic decrease of resistance to impact by a factor of 10). The transition of gelatin to the rubbery state at P/P0>O.8 is conformed by variations of resistance to impact of hot gelatin films. As can be seen from Figure 4 (curve 1), resistance to impact of hot films also increases (by a factor of about 5)in this moisture content region. It is of interest that over the whole region of the glassy state, the resistance to impact of coiled gelatin films is practically unaffected by variations of the moisture content, and the resistance to impact is well below that for helical gelatin by its absolute value. The above findings show unambiguously that water is not a mere plasticizer for gelatin. Rather, its influence on the gelatin properties in the glassy state has a strong bearing on the gelatin structure, viz. the presence of collagen-like helical formations. Indeed, there is a good correlation between the moisture-content dependences for (i) the resistance to impact of helical gelatin (Figure 3), (ii) the second moment of the n.m.r. absorption line and (iii) the long transversal relaxation time (Figure 5)90.
Investigations into the stability of the solid gelatin structure and of the structural transformations occurring in gelatin during heating or cooling have grown of importance because of the use of gelatin over a wide temperature range.
Figure 6. Dilatometric curve for a cold gelatin film
Linear dilatometric studies of cold gelatin films at.-100° to 230°C have revealed the existence of three temperature regions differentiated by the type of solid gelatin behaviour displayed. (Figure 6) 29,115
Figure 7 Dilatometric curves for cold (a) and hot (b) gelatin films kept for 2 weeks at the air humidities of 0 (1); 10 (2); 20 (3);40 (4); 65 (5);and 80% (6)
In the first region (-100° to 20°C) gelatin exhibits reversible expansion and shows a number of temperature transitions on the dilatometric curves, because the gelatin-water system is in a non-equilibrium state116, The behaviour of gelatin in this temperature region is essentially the same as that of hydrophilic polymers with positive linear expansion coefficients typical of them.
In the second region (20° to 120°C) the behaviour of gelatin is mainly determined by its moisture content. The thermal contraction of cold and hot gelatin films observed in this temperature region (Figure 7) is not associated with relaxation processes since it is independent of the conformational state of gelatin macromolecules. Rather, it is in essence a purely physical contraction of samples caused by water desorption. This contraction is completely reversible on repeated moisturing of the gelatin116-120 and correlates well with the contraction caused by decreasing air humidity at room temperature121.
Thermal treatment of oriented cold gelatin films results in a contraction which shows anisotropy along and across the direction of stretching. Increase in the degree of ordering in gelatin leads to decrease in the longitudinal and an increase in transversal contractions122.
Figure 8 Dilatometric curves and supercontractions (a and b, respectively) of gelatin films cast at various temperatures ((1) 6°C; (2) 20°C; (3) 45°C; (4) 60°C)
Figure 9 Thermorelaxational and thermomechanical (2,3) curves for cold gelatin films. Load: 380 g mm-2 (2) and 285 g mm-2 (3). (1,2) not annealed films; (3) films annealed at 190°C
The third region (120° to 225°C) comprises two subregions, the pre-transitional one (120° to 200°C) and the temperature region (200° to 225°C) of the helix-coil conformational transformation of solid gelatin macromolecules. The transformation is followed by the appearance of rubbery properties in the gelatin116,123,124, En the pretransitional subregion (120° to 200°C) gelatin also exhibits contraction caused by both the removal of the remaining water and by a thermal relaxation process induced by the increase in mobility of the individual structure elements of gelatin. Macroscopically, the helix to coil conformational transition occurring in solid gelatin shows itself as an irreversible spontaneous sample supercontraction (of up to 30% of the initial sample length) at approximately 210°C. Supercontraction of gelatin is similar to that observed in collagen. Its magnitude depends directly on the initial conformational state of the gelatin macromolecules (Figure 8). The film thickness increases by a factor of 2 to 3 upon supercontraction. The X-ray diffraction patterns of the films taken after the supercontraction show the disappearance of the 2.8, 7.5 and 11.4 Å spacings indicative of the loss of helicity Of gelatin macromolecules123. Figure 8b shows the dependence of supercontraction of gelatin films on the casting temperature. The linearity observed suggests that the degree . of uncoiling (anisotropy) of gelatin macromolecules can be inferred from the magnitude of the thermal supercontraction. The conformational transition of gelatin occurs with heat consumption (l7cal/g27, 6.4cal/g125) and hence exhibits an endothermic peak on d.t.a. curves119. Besides, supercontraction is independent of the initial moisture content of gelatin116, of planar orientation of the structural elements120 but decreases with increasing external, load124. A critical load (~500 g mm-2) has been found at which gelatin undergoes elongation with heating and shows no super-contraction effect124. A suggestion has been made that gelatin macromolecules tend to coil after thermal break of bonds, stabilizing the helical conformation with the force equal to that of the critical load,
The supercontraction temperature of gelatin, Ts, is independent of the conformational state of its macromolecules116,123 and of the load applied124 but is dependent on the rate of heating.
The temperature of the transition appearing on the thermomechanical curves of cold films (Figure 9, curves 2 and 3) coincides with Ts (curve 1). Gelatin also shows a sharp drop in relative rigidity at 200°-210°C which coincides with the maximum of the mechanical damping index126.
Figure 10 Dilatometric curves for a cold gelatin film on heating under the load of 160 (1)and 425 (2) g mm-2 and on cooling after the removal of load (1', 2')
When Ts (2 10°C) is attained, gelatin turns rubbery and is characterized by large reversible deformations (up to 100% and more) at comparatively low loads. It can be seen from Figure 10 that on cooling and removal of load, gelatin samples contract sharply to regain their initial length within a narrow temperature range (2°-3°C). Thermomechanical studies of gelatin, subjected to thermal supercontraction, have shown that the only sharp temperature transition (204°-209°C) in this coiled gelatin occurs at the temperature coincident with 7 for helical gelatin (205°-210°C). After coiling and removal of load, stretched coiled gelatin films exhibit reversible contraction which is essentially completed at 213°C124. Consequently, Ts of helical gelatin coincides with Ts of coiled gelatin.
Proceeding from experimental evidence available at present, the gelatin supercontraction process may be thought to comprise of at least two steps. The first step is (at ~200°C) degradation of the gelatin helical structure which occurs with the breaking of three-stranded segments into separate chains (X-ray diffraction patterns only show a diffuse halo116,124). Gelatin macromolecules acquire high mobility causing subsequent transition of macromolecules to a statistical coil state (at ~210°C) and supercontraction of the sample.
When heated, gelatin undergoes not only structural and mechanical but also, physico-chemical transformations116,119 such as partial or complete loss of solubility in water caused by crosslinking and lowering of molecular weight caused by thermal and thermo-oxidative destruction. These changes are observed in heating gelatin above 140°C Above 170°C, gelatin is insoluble in hot water116.127. Thermally treated gelatin occurs in water as an elastic gel-like film. The formation of chemical bonds between gelatin macromolecules in heating above 140°C is confirmed by its insolubility in hot saturated urea solutions116. As soon as gelatin undergoes transformation to the rubbery state (at ~210°C) and its viscosity decreases, its liability to thermal destruction and, in the presence of air, thermo-oxidative destruction increases.
It therefore follows that the thermal stability of gelatin, determined by the temperature at which it begins to decompose, is close to its heat resistance determined by the supercontraction temperature.
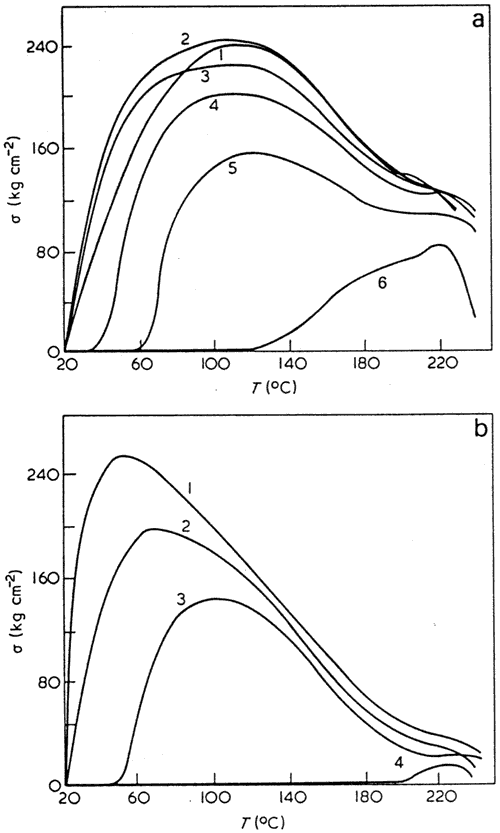
Figure 11 Stretching diagrams for cold gelatin films (a) at 20 (1), 70 (2), 100 (3), 150 (4), and 220°C (5) and the temperature dependences of ultimate stress (1b) and deformation 2b)
Throughout the temperature range of the glassy state (20° to 220°C) gelatin behaves as a brittle solid with a very low deformability (3-7%). Stretching diagrams of gelatin films at various temperatures shown in Figure 11 provide a good illustration of this129.
The brittle fracture is also typical of air-dry gelatin films (at 20°C, curve 1). The film strength, however, decreases sharply (nearly twofold) if gelatin macromolecules change their conformational state (curve 5). The most sensitive characteristic is the resistance of films to impact. This decreases from 70 kg cm cm3 at 20°C to 25 kg cm cm3 at 70°C (i.e. about threefold). Heating of gelatin containing a large number of functional groups increases intermolecular interactions and hence material rigidity. This effect outweighs the increase in thermal mobility of structural fragments. Moreover, even the transition of gelatin to the rubbery state does not increase its elasticity because it coincides with the helix to statistical coil conformational transformation resulting in the loss of all useful mechanical properties of polymers.
The internal stress in gelatin materials heated .under isometric conditions has been studied in some detail130,131. Figure 12 shows the temperature dependence of internal stress in air-dry (65 r.h., curve 1) and dry (0% r.h., curve 2) cold gelatin films. As can be seen from Figure 12, air-dry gelatin features a significantly different temperature dependence from dry gelatin which suggests different stress origins. A study of various factors affecting internal stress over a wide temperature range (20° to 240°C) has shown that there are three temperature ranges (A, B and C) differing by the gelatin behaviour130, These ranges correlate well with those observed for gelatin dilatometric curves (ranges II, III and III', Figure 6).
Figure 12 Temperature dependence of internal stress in air-dry (1) and dry (2) cold gelatin films
In temperature range A, (20° to 120°C) gelatin films show large internal stresses resulting from gelatin contraction caused by water desorption (Figure 12 curve '1) In dry gelatin these stresses are absent (Figure 12 curve 2). The upper temperature limit of this range is determined mainly by the conformational state of the gelatin macromolecules and consequently by the cohesion strength of the gelatin. For helical gelatin this upper limit is 120°C while for coiled gelatin it is shifted to about 50°C (Figures. 13 and 14) because of the earlier appearance and development of cracks' 30, To large part stress increases during the first 10-15 mm of heating after the loss of 0.5 to 1.2% water (to more than 200 kg cm-2)130.
Figure 13 Temperature dependence of internal stress in cold (a) and hot (b) gelatin films kept at water vapour pressures of 0.8 (1); 0.65 (2); 0.4 (3);0.2 (4);0.1 (5);0 (6)
Figure 14 Temperature dependence of internal stress in gelatin films cast at 4 (1, 11), 20 (2, 2'). 25 (3, 3'), 30 (4,4'), 35 (5, 5'), 40 (6, 6') and 60°C (7, 7'). (1-7) air-dry, (1'-7') dry films
In temperature range B (120° to 220°C) additional stress associated with relaxation processes and resulting from film shrinkage develops in gelatin116 (Figure 12, curve 2). This magnitude and the temperature at which this stress appears depends on the conformational state of the gelatin macromolecules (Figure 14, curves 1'-7').
In temperature range C, above 220°C (Figure 12), a sharp acceleration of the processes associated with stress relaxation is observed regardless of the conformational state of the gelatin macromolecules and the initial moisture content (Figures 13 and 14). This acceleration results from the transition of gelatin to the viscous-flow state119.
The experimental data described above suggest that isometric heating of gelatin, which is a constituent of many materials used in the production of various articles, results in extremely high internal stress which can be on a par with gelatin strength. This is, e.g. the case with photographic materials where gelatin layers are tightly bound to backings. If gelatin materials are kept under isometric conditions for a sufficiently long period of time this cannot but lead to prematory breakdown of articles made of these materials.
Figure 15 Stress relaxation in air-dry (a) and dry (b) cold gelatin films at 20 (11:30 (2); 50 (3); 70 (4); 100 (5); 150 (6); 180 (7);200 (8) and 220°C (9). (a') curve 7 scaled up; (c) the temperature dependence of the stress relaxation rate
The stress relaxation curves for air-dry (65% r.h.) and dry (c.a. 0% r.h.) cold gelatin films over a wide temperature range taken under isometric and isothermal conditions are shown in Figure 15132, As can be seen from Figure 15, the shape of the relaxation curves and the relaxation rate strongly depend on the moisture content. With air-dry gelatin, relaxation processes of two types are distinctly observed: fast and slow relaxation processes (Figure 15a). The fast relaxation features an anomaly at temperatures above 100°C which is not observed with synthetic polymers, viz. the fast relaxation (taking about 5 seconds, Figure 15a') is replaced by the slow relaxation after a spontaneous increase of stress. At the same time, no increase in stress is observed with dry gelatin films (Figure 15b). This suggests that the origin of the anomaly is the presence of water in the air-dry gelatin. Consideration of the relaxation curves in Figure 15a shows that after the load is removed, two relaxation processes occur in gelatin: almost instantaneous elastic stress relaxation and increase of stress caused by unrealized gelatin contraction. The two processes have opposite effects and therefore compensate each other. Stress relaxation in dry gelatin also shows some specific features (Figure 15b). At 20° and 30°C the stress gradually decreases to zero for about 50 min (curves 1, 2). At higher temperatures the stress does not disappear completely but the relaxation rate rises appreciably (Figure 15c). Under heating, two processes occur in dry gelatin: water sorption from the surrounding medium and water desorption caused by gelatin heating. At 20° and 30°C, water sorption only takes place, and the mobility of all structural elements increases appreciably to provide complete relaxation of. stress, In this case the stress relaxation rate only depends on the relative pressure of water vapour. The relaxation rate increases substantially with air humidity to the extent that relaxation occurs almost instantaneously133,134. At higher temperatures above 30°C the rate of desorption exceeds the rate of water sorption and the amount of water sorbed is insufficient to provide mobility of gelatin structural elements needed to achieve complete relaxation.
The studies of mechanical relaxation processes thus reveal the rigid-chain nature of this polymer and the role played by water in its stress relaxation. The data discussed above suggest that the relaxation processes observed in heated gelatin involve only the elastic component of deformation.
Physical modification of the properties of polymeric materials implies an alteration of the molecular and supramolecular structure of a polymer during its processing. Structural modification may involve changes of Conformations of polymer macromolecules, orientational changes at the molecular and supramolecular levels and changes of the phase state.
Physical modification of gelatin includes two groups of methods: renaturation of the collagen-like helical structure and orientation of gelatin macromolecules.
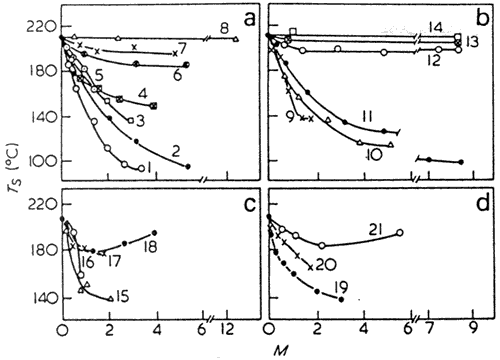
Figure 16 Dependence of Ts of cold gelatin films on the number of moles of low-molecular weight compounds introduced into the films: (a) 1, triethanolamine; 2, glycerin; 3, resorcin; 4, thiodiglycol; 5, sodium salt of toluenesulphoacid; 6, 1,4 butylene glycol; 7, ethylene glycol; 8, ethylenechlorohydrin. (b) 9, urea nitrate; 10, thiourea; 11, urea; 12, dimethyl sulphoxide; 13, acetamide; 14, formamide. (c) 15, glutamic acid; 16, aspargic acid; 17, valine; 18, glycine. (d) 19, KSCN; 20, KI; 21, LiBr
Conformational methods of modification. Controlling the processes of conformational transformations is known to be a very convenient means for manufacture of polymeric materials135. Gelatin processing may serve as a good example of the production of polymer materials with specifically intended conformations of macromolecules. At elevated temperatures gelatin molecules are known to form coils in water which undergo transformation to give collagen-like helical formations during casting at reduced temperatures. Thus the modification technique only involves cooling of the solution and storage of the resulting gel for some time in order that the molecules attain the highest degree of helicity, This technique, however, falls behind the demands of large-scale production of gelatin materials. Up-to-date polymer industry requires intended renaturation of gelatin involving no cooling (gel formation) stage. This problem, though of great practical and theoretical interest, is still far from being resolved.
Orientational methods of modification. The second group of physical methods used for modification of gelatin involves orientational stretching to obtain gelatin fibres or other materials possessing high strength along the stretch direction136,137. Bergmann and Jacobi138 were probably the first to investigate the strength of stretched gelatin and to show that this material had a tensile strength of 9.3 kg mm-2 along the stretching axis and 4 kg mm-2 in the direction normal to that axis. Even stronger gelatin fibres were obtained by Anokhin136, by casting fibres from 60% gelatin solutions and applying fourfold stretching. His fibres showed high strengths, for such materials, ranging from 10 to 20 kg mm-2 These few investigations were made to develop the technique for manufacturing fibres for specific, e.g. medical, purposes. Such fibres, however, have found no practical application unlike other protein fibres (casein, vegetable proteins, collagen fibres) which are produced commercially, but only on a limited scale.
Physico-chemical modification of polymeric substances implies the preparation of two- or multi-component systems modifying the chemical composition and structure of the material, but involving no chemical reaction between the components. Physico-chemical modification includes the use of various methods for plastization of polymers and for construction of heterogeneous, systems.
We will consider these two groups of methods in more detail.
Plasticizing of gelatin. Like other polymers gelatin is plasticized to increase its elasticity, decrease transition temperatures and modify its 'other physico-chemical characteristics. However, in some cases the behaviour of gelatin differs markedly from that of other polymer glasses139. This mainly refers to the effects of plasticizers on gelatin transition temperatures and mechanical properties, in particular, on gelatin resistance to impact. Most plasticizers reduce gelatin's Ts (Figure 16)65,140,141, Decrease of Ts however, depends appreciably on the chemical structure and size of the low-molecular weight compounds introduced into gelatin65,140.
Figure 17 Dependence of contraction of cold gelatin films in the range 20°C to 120°C on the number of moles of low molecular weight substances introduced into the films (the designations are the same as in Figure 16)
Effects of low-molecular weight compounds on thermal contraction of gelatin below 120°C, i.e. contraction caused by water desorption116 are similar to their effects on the Ts. As can be seen from Figure 17, the concentration dependences of the thermal contraction and 7; are identical except for a few cases (cf Figure 16). This is rather interesting since the two processes differ in nature.
Figure 18 Dependence of resistance to impact on the water vapour pressure for cold gelatin films containing: (a) glycerin (0 (1). 10 (2), 20 (3), and 30% (4)); (b) urea (0 (1), 5 (2), 10 (3), and 20% (4)); (c) ethylene glycol (0 (1) and 30% (2)) and triethanolamine (20% (3))
Recent detailed studies of resistance to impact and sorption properties of gelatin films containing plasticizers and free water gave the lowest prerequisite moisture content for a plasticizer to have a modifying action142, As can be seen from Figure 18 the resistance to impact dependence of the plastized gelatin films on their moisture contents always exhibit maxima. Whatever the type and concentration of the low-molecular weight compound and its water sorption ability, the maximum occurs in the same region (P/P0 ~.0.5-0.7). The maxima are associated with the transition of the gelatin to the rubbery state. As can be seen from Figure 18, neither of the low-molecular weight compounds studied has a plasticizing action at P/P0 <0.5-0.6, i.e. resistance to impact of films containing these compounds is lower than or, at best, the same as that of gelatin films free of plasticizers. And it is only at a definite moisture content (~12% water) that low-molecular weight compounds have a plasticizing action and the pattern typical of plasticizing of polymers is observed. Interestingly, this minimum percentage of water is independent of the nature of the low-molecular weight compound introduced into the gelatin. None of the low-molecular weight substances studied has a modifying action on anhydrous gelatin. It should, however, be noted that low-molecular weight compounds appreciably reduce internal stress in both air-dry and dry gelatin films subjected to isometric heating143.
Figure 19 Dependence of impact strength on the pressure of water vapours for cold gelatin films containing: (a) PEG-300--0 (1), 5 (2), 10 (3), 20 (4) and 30% (5); (b) PEG-3000-0 (1), 1(2), 3 (3) and 5% (4); (c) PEG-40000-0 (1), 0.1 (2), and 1% (3)
Modification of gelatin in formation of heterophase systems. Modification of gelatin with high-molecular weight compounds may be accomplished by the introduction of hydrophilic or hydrophobic polymers (in the form of latexes). Modification of gelatin with hydrophilic polymers was thoroughly studied with polyethyleneglycols (PEG) which form mixtures with gelatin of limited miscibility142-144: As can be seen from Figure 19, resistance to impact of gelatin film, containing PEG as a function of water vapour pressure, features a maximum. The position of the maximum depends on the molecular weight and concentration of PEG and corresponds. to the optimum degree of microdemixing of the system. As the moisture content increases (P/P0 >0.7) the system undergoes macrodemixing which manifests itself by turbidity and lowering of resistance to impact. Increase of resistance to impact of gelatin films is also observed with other hydrophilic (PVC, agar) and hydrophobic polymers as admixtures145, In this case the concentration dependence of resistance to impact also exhibits a maximum whose height and position depend on the chemical nature145 and molecular weight144 of the polymer. The thermophysical properties of gelatin- polymer systems such as thermal contraction and heat resistance temperature, Ts are close to those of unmodified gelatin144,146. Hydrophilic oligomers (for instance, PEG-300, PEG-400144) like low-molecular weight compounds have a stronger effect on those characteristics. The effect of PEG of various molecular weights on the appearance, growth and relaxation of stress in gelatin films during their isometric heating has been studied143 and shown that PEG, although it reduces stress in air-dry gelatin films, does not inhibit the displaying of the intrinsic gelatin matrix properties.
Figure 20 Dependences of the limiting shear stress of gelatin gels (a) and the impact strength (P) of gelatin films (b) on DMSO content in the initial gelatin solutions. (a) gelation time: 60 (1), 75 (2), 120 mm (3), 1 day (4), and 4 days (5); (b) P/P0: 0 (11,0.3 (2), 0.65 (3), 0.8 (4) and 0.9 (5)
In discussing the physico-mechanical properties of gelatin-based heterophase systems it seems pertinent to mention one more example, viz. heterophase systems containing a single polymeric component (gelatin) and formed in coacervation of gelatin by addition of certain organic solvents to its aqueous solutions. Two liquid phases present in these systems contain gelatin in different concentrations. One of the phases (the more concentrated one) forms Coacervative drops' which act as a disperse phase147. The limiting shear stress of gels and resistance to impact of films made from aqueous gelatin solutions containing DMSO exhibit maxima at 50 vol% DMSO (Figure 20). As the DMSO concentration increases macrodemixing of the system with the appearance of sharp interphases occurs. The gels and films turn white and their strengths decrease.
To sum up, non-modified gelatin undergoes brittle fracture under impact even at ordinary moisture contents. Increase of resistance to impact is only observed when gelatin undergoes transformation to the rubbery state or occurs as constituent in heterophase systems with the latter seeming to offer more promise. The microheterophase systems show less severe humidity dependences than nonmodified gelatin.
The methods of chemically modifying gelatin take advantage of the presence of various functions in gelatin that are reactive towards various low- and high-molecular weight compounds. These methods will be subdivided into three groups: (1) polymer-analogous conversions; (2) crosslinking; (3) graft copolymerization.
Polymer-analogous conversions. These methods were employed both for purely scientific purposes and to improve some technological processes for manufacturing new materials with the desired properties. The scientific utility of these methods comes mainly from the possibility they offer for elucidating the mechanism and nature of gel formation in gelatin148-151. As for technological applications, gelatin thus modified is widely used in the manufacture of photographic emulsions by a solid phase deposition method152,153, in microcapsulation of hydrophobic substances154, in medicine155 and various industries.
Crosslinking. Chemical modification by crosslinking should directly affect the physico-mechanical properties of gelatin. These methods are widely used in the manufacture of various gelatin and collagen products and seem to offer limitless possibilities for modifying the properties of gelatin because the number of bi- and polyfunctional organic and inorganic compounds that can interact with the particular gelatin functions is very large indeed. Crosslinking of gelatin macromolecules is known to increase the viscosity of the gelatin solutions, strength and melting points of gelatin gels, and to affect all the physicochemical and mechanical properties of solid gelatin. A linear relationship between the swelling and rigidity modulus of hardened gelatin layers independent of the nature of the hardener, the temperature and the conformational state of the gelatin macromolecules has been observed156 It is noteworthy that crosslinking of gelatin has no effect on the helix formation in solutions60,61. As for solid gelatin, the crosslinking has practically no effect on its heat resistance (Ts) but substantially reduces its supercontraction141. It is also of interest that the presence of hardener does not hinder the decrease of Ts under the action of plasticizers157.
Figure 21 Stretching diagrams for films made of gelatin (1) and of graft copolymers of gelatin with 6 (2), 27 (3), 40 (4), 60% (5) PBAC and 3(6), 46% (7) PAN164
Graft copolymerization. The third group of methods employed for chemically modifying gelatin is the preparation of graft copolymers based on gelatin and synthetic polymers. The synthesis, structure, thermophysical and physico-mechanical properties of graft gelatin copolymers have been studied in detail58-164 It is important that graft gelatin copolymers retain the valuable properties of the parent gelatin: the ability to form gels and helices and the high heat resistance. At the same time, gelatin acquires new properties depending on the .nature of the grafted polymer. Thus grafting of rubber-like polybutylacrylate (PBAC) and rigid-chain polyacrylonitrile (PAN) onto gelatin yields products differing greatly in their properties (Figure 21). The dependence of the physico-mechanical properties of graft gelatin copolymers on the conformational state of gelatin macromolecules has attracted much interest of researchers163. The grafting of PBAC, a flexible-chain polymer, onto coiled gelatin produced a substantial increase in film elasticity (Table 5). As can be seen from the data in Table 5, PBAC is capable of improving the mechanical properties of coiled gelatin to the extent that the product compares well with helical gelatin, which cannot be done otherwise, and even graft helical gelatin.
The analysis of the structure and properties of gelatin in the solid state over a wide temperature and humidity range has revealed the specific' features of gelatin responsible for all its physico-mechanical properties. Firstly, gelatin behaves very differently depending on whether its macromolecules are in the collagen-like helical or coiled conformation. In the former case articles made of gelatin exhibit properties suitable for service at ordinary temperatures and humidities, while in the latter case gelatin behaves as a brittle impractical material.
Secondly, gelatin is a typical rigid-chain polymer. Because of this gelatin behaves as a brittle material regardless of molecular conformations in the absence of a solvent (water).
Thirdly, gelatin features a rather wide temperature range of the glassy state with the upper limit of 205°C-210°C. This characterizes gelatin as a material with a fairly wide range of heat resistance.
Finally, gelatin as a hydrophilic biopolymer specifically interacts with water and undergoes drastic changes of its physico-mechanical properties depending on the moisture content.
It thus follows that the manufacture of gelatin articles intended for use in wide temperature and humidity ranges requires some modification of gelatin. We would like to focus the attention of researchers on two fundamental problems which still remain to be solved. These are the 'conformational' problem and the problem of gelatin plasticizing.
Let us consider the 'conformational' problem first. The technological principles of manufacturing articles having suitable mechanical properties are as follows:
(1) the temperature of gelatin solutions to be used for film or layer casting should provide the most compact state of gelatin macromolecules, i.e. the temperature should be above 45°C when the solution is most mobile;
(2) the temperature and duration of film casting should provide the optimum conditions for formation of the spatial structure of gel and attainment of the highest degree of renaturation of the collagen-like helical structures possible (<20°C).
The second requirement is contradictory to the current tendency to intensify technological processes. In view of this, systematic studies in two principal directions are urgent:
(a) The search for specific structurizers accelerating formation of the collagen-like helical structure in gelatin rather than gelation of gelatin solutions (substances accelerating the gelation are available). The introduction of such structurizers would make it possible to obviate the necessity of solution gelation. At present, gelation is the only route to helical gelatin.
(b) The search for ways to improve the physico. mechanical characteristics of gelatin with a globular supramolecular structure. This can probably be accomplished by chemical modification, such as graft copolymerization, of synthetic flexible-chain polymers with gelatin.
The second problem is the problem of gelatin plasticizing. When gelatin materials are used at ordinary temperatures and humidities and when the fibrillar structure and the necessary moisture content are retained plasticizing of gelatin poses no problem. If however these conditions are not met the usual plasticizing methods fail. In this case the problem can be solved, in our opinion, by modification of gelatin with high-molecular weight compounds, viz. by the preparation of heterophase systems featuring less severe humidity dependences than the parent polymer. Such modification of gelatin layers is now finding ever increasing applications and is carried out either by introducing latexes into the gelatin layers or by grafting flexible-chain polymers onto gelatin macromolecules as there seems to be no other method available.
1. Sokolov, S.I. 'Phisical Chemistry of Collagen and its Derivatives', Gizlegprom, Moscow-Leningrad, 1937
2. Zaides, A.L. 'The Structure of Collagen and its Changes in 1960
3. Adv. Protein Chem., Academic Press, Processing', Legkaya Insustriya, Moscow, 1971
4. Mikhailov, A.N. 'Collagen of Skin and Principles of its Processing', Legkaya Insustriya, Moscow, 1971.
5. Raikh, G. Kollagen (Collagen), Legkaya Industriya. Moscow, 1969
6. Chien, J.C. W. Macromol, Sci., Rev. Macromol. Chem. 1975 C12(1), 1
7. Veis. A, 'The macromolecular Chemistry of Gelatin', Academic Press, New York, 1964
8. 'Recent Advances in Gelatin and Glue Research', (Ed. Y. Stainsby), Pergamon Press, London and New York, 1958
9. Yannas, 1. V. J. Macromol, Sci., Rev. Macromol. Chem. C7(l), 49
10. Hopp, V. Chemiker - Ztg. Chem. Apparature 1965, 89, 469
11. Hill, T.T., Adv. Chem. N 78, 381
12. Miz, K., and James, T., 'Theory of Photographic Process', Khimiya, Leningrad, 1973
13. Brezlav,Yu, A.Z. Nauchnoi i Prikladnoi Fotografii i Kinematographii, 15, 458
14. Hill, T.T. Literature on Gelatin. Chapter in Literature Resources of the Chemical Process Industries, 2nd Edn., vol. 1, American Chem. Soc., Washington, 1965
15. Tucker, H. A. 'Absorbable gelatin (gelform) sponge. An annotated bibliography', 1945-1965, C. C. Thomas, 111, 1965
16. Titov, A. A. 'Proteins in Industry and Agriculture', Academy of Sciences of the USSR, 1952, p.162
17. Wood, H.W. J. Photogr. Sci. 1961,9, 151
18. Borginon, H. J. Photogr. Sci. 1967, 15, 207
19. Zubov, P.I. Moscow, 1947
20. Ferry, J.D. Adv. Protein Chem. 1948. 4,2
21. Ward, A.Y. Research 1954, 5, 85
22. Ward, A.Y. Research 1951, 4, 119
23.. Rogovina, L.Z. and Slonimskii, G.L. Uspekhi Khim. 1974, 43, 1102
24. Papkov, S. P. 'Gel State of Polymers', Khimiya, Moscow, 1974
25. Izmailova, V. N. and P. A. 'Structure Formation in Protein Systems', Moscow, 1974
26. Irie. H. J. Soc. Photogr. Sci. Technol. Jpn. 1977, 40, 130
27. Jolly, I.E. Photogr. Sci. Eng. 1970, 14, 169
28. Kozlov, P.V., Burdygina, G.I., Fridman, I.M., Moteneva, Zh. F. and Yarysheva, L.M.Z, Nauchnoi I Prikladnoi Fotographii i Kinematographii 1972, 17, 59, 228
29. Kozlov, P.V. and Burdygina, G.I. Z. Nauchnoi I Prikladnoi Fotografii i Kinematographii 1977, 22, 68
30. Kargin, V.A, Kozlov, P.V. and Undzenas, A.I. Uspekhi Nauchnoi Fotografii 1972,16, 122
31. Morawetz, H. Adv. Protein Chem. Academic Press, London and New York, 1972, 26, 243
32. Flory, P.J., and Weaver, E.S. J. Am. Chem. Soc. 33, 1960, 82, 4518
33. Pouradier, J. and Venet, A. M. J. Chim,. Phys. Chim., biol. 1950, 47, 11, 391; 1952, 49, 85, 238; 1961, 58, 778
34. Zubov, P.I., Zhurkina, Z. N. and Kargin, V.A. Dokl. Akad. Nauk SSSR 1949, 62, 659
35. Kargin, V. A. Uspekhii Khim 1966, 35, 1006
36. Kapralova, Z. A., Mirlina, S. Ya., Kozlov, P. Popova, L.A. Vysokomol. Soedin. 1962, 4, 321
37. Rice, R. V. Proc. Nat. Acad. Sci., USA. 46, 1186
38. Slonimskii, G. L., Kitaigorodskii, A.I., Belavtseva, E.M., Tolstoguzov, V. 1. Vysokomol. Soedin. 1968, B10, 640
39. Papkov, S.P., Shchetnev, Yu. F, Iovleva. M.M. and Banduryan, S.I. Vysokomol. Soedin. 197!, 813, 720
40. Belavtseva, E.M., and Titova, E.F. Vysokomol. Soedin. 1972, A14, 1659
41. Belavtseva, E.M., Titova, E.F., Braudo, E. and Tolstoguzov, V.B. Biofizika 1973, 18, 929; 1974. 19, 19
42. Mikhailov, A. N., Titova, 8. F. and Belavtseva, E. M. Biofizika 1979, 24, 438
43. Courts, A. and Stainsby, C. 'Recent Advances. Gelatin and Glue 1958. p. 100
44. Hippel, P.H. and Wong, K.Y. Biochemistry, 1963, 2 1399
45. Harrington, W. F. and Hippel, P. H. Arch, Biochem. Biophys. 1961, 92, 100
46. Merzlov, V.P. and Pchelin, V.A. Akad. Nauk SSSR 1965, 163, 147
47 Boedker, H. and Doty, P. J. Phys. Chem. 1954, 58, 968
48 Katz, 1. 'X-Ray Diffraction by Colloids and Tissues', ONTI, 1937
49. Zaides, A. L. and Sokolov, S. I. 'The Structure and Physico Mechanical Properties of Rubber, Collagen and Cellulose Derivatives', Legkaya Promyshlennost', 1937, p 141
50. Robinson, C. and Bott., M.J. Nature 1951, 168, 325
51. Bradbury, E. M. and Martin, C. Proc. Roy. Soc., London A214, 183
52. Ramachandran, G. N. 'Recent Advances in-Gelatin and Glue Research', (Ed. G. Stainsby), Pergamon Press, London and New York, 1957, p 32
53. Robinson, C. 'Nature and Structure of Collagen', London, 1953. p 96
54. Coopes, I.H. J. Polym. Sci. 4-7 1968, 6, 1991; 1971, 9, 3683
55. Coopes, I.H. J. Photogr. Sci. 1974, 222,269
56. Johnson, M.F., Fellows, W.D., Kamm, W.D., Miller, R.S. Otto, H.O. and Curme, H.G. 'Photographic Gelatin',(Ed. R. C. Cox), Academic Press, London, 1972, p 99
57. Nagao, K. 'Photographic Binders', (Ed. H. Irie et al. Japan, 1976, p.1
58. Kozlov, P.V., Undzenas, A.I., Merzlov, V.P. and Rozenberg, S.G.. Dokl. Akad. Nauk SSSR 1969, 185, 118
59. Rogovina, L.Z., Slonimskii, G.L. and Aksenova, P.L. Vysokomol Soedin, 1971, A13, 1451
60. Coopes, I.H. J. Polym. Sci. A-1, 1970, 8, 1793
61. Moll, Fl, Rosenkranz, H., and Himmelmann, W. J. Photogr. Sci. 1974, 22, 255
62. Askadskii, A.A. 'Physical Chemistry of Polyarylates', Khimiya, Moscow, 1968
63. Pchelin, V. A. Dokl Akad. Nauk SSSR 1968, 180, 402
64. Yannas, I. V. and Huang, C. Macromolecules 1972, 5, 99
65. Moteneva, Zh. F., Burdygina, G. 1., Fridman, I. M. and Kozlov, P. V. Vysokomol. Soedin. 1974, A16, 1113
66. Burdygina, G.I. Mokina, N. V. and Kozlov, P.V. Vysokomol. Soedin, A19, 2132
67. Pchelin, V.A. and Nikolaeva, S.S. 'Uspekhi Kolloidnoi Khimii, Nauka', Moscow, 1973, p 357
68. Kogure, M., Hogi, T., Tamura, M., Ogi, K. and Nakadate, T. 'Photographic gelatin II', (Ed. R. J. Cox), Academic Press, London and New York, 1976, p 131
69. Kuntz, I.D. and Kauzmann, W. 'Advances in Protein Chemistry', Academic Press, New York and London, 1974, 28, 239
70. Prialov, P.L. Biofizika 1968, 13, 163
71. Askochenskaya, N. A. and Petinov, N.. Uspekhi sovremennoi Biologii 1972, 73, 288
72. Bull, M. B. J. Am. Chem, Soc. 1944, 66, 1499
73. Pouradier, J. J. Chem. phys. et Phys.-chim. biol. 1970, 67, 229
74. Burdygina C.I., Moteneva, Zh. F., Fainberg, E. Z. and Kozlov, Dokl. Akad. SSSR 1976, 231, 116
75. Iwamoto, K. and Fujii, T. 'Photographic Binders', (Ed. H. Erie et al). Japan, 1976, p 47
76. Papkov, S. 'P. and Fainberg, E. Z. 'Interaction to Cellulose and Cellulose Materials with Water', Khimiya, Moscow, 1976
77. Brunauer, S., Emmett, P. H. and Teller, E. J. Am. Chem. Soc. 1938, 60, 309
78. Pasynskii, A.C. and E1'piner, I.E. Akad. Nauk SSSR 1955, 105, 1296
79. Masuzawa, M. and Sterling, C. J. Appl. Polym. Sci. 1968, 12, 2023
80. Eliassaf, J., and Errich, F.R. J. Appl. Polym. Sci. 1960, 4, 200
81. Burdygina C.I., Chenborisova, L. Ya., Maklakov, A.I. and Kozlov, P.V. International Symposium on Macromolecular Chemistry, Taskent, Abstracts, 1978, 5, 170
82. Chenborisova, L. Ya., Burdygina, G. I., Maklakov, A.1. and Kozlov, P. V. Dokl. Akad. Nauk SSSR 1979, 248, 154
83. Guillet, J., Seytre, C., May, and Vallot, G. Polym. J. 1975,7, 26
84. Pasynskii, A. C. Kolloid. Z. 1946, 8, 53
85. Grollmann, A. J. Gen. Physiol. 1932, 14, 661
86. Dumanskii,A. V. and Nekrich, E. F. Kolloid. Z. 1955,17,168,173
87. Vinetskaya, E. Ya. Proceedings of TsNIKP, No. 2, 77, 1934
88. Kuntz, I.D. J. Am. Client, Soc. 1971, 93, 514
89. Kuntz, 1.D., Brassfield, T. S., Law, C. and Purcell, C. Science 1969, 163, 1329
90. Chenborisova, L. Ya., Burdygina, C. I., Maklakov, A. I. and Kozlov, P. V. Vysokomol. Soedin. 1978, A20, 2838
91. Sorokin, G.A. Vysokomol. Soedin. 1971, A13, 608
92. Dowell, L.G., Moline, S.W. and Rinfret, A. P. Biochim. Biophys. Acta 1962, 59, 158
93. Moran, T. Proc. Roy. Soc., London 1930. A 112, 30; 1932, A 135, 411; 1930, B107
94. Moran, 1. Kolloid-Z. 1932, 59, 217
95. Privalov, P.L. and Mreviishvili, C.M. Biofizika 1967, 12, 122
96. Esipova, N.C., Andreeva, N.S. and Gatovskaya, T.V. Biophizika 1958, 3, 529
97. Dehl, R.E. Science 1970, 170, 738
98. Fung, B. M. and Wei, S. C. Biopolymers 1973, 12, 1053
99. Hal6 (sic), A. R. and Snaith, J. W. Biopolymers 1971, 10, 1681
100. Cheshire, A. and Holmes, N.J. Soc. Leather Trad. Chem. 1942, 26, 237
101. Nomura, S., Hilther, A., Lando, J.B. and Baer, E. Biopolymers 1977, 16, 231
102. Berendsen, J.C. J. Chem. Phys. 1973, 59, 296
103. Dumanskii, A. V. and Nekrich, E. F. Ukrain. Khim. Zh. 1960,26, 289
104. Shiraishi, H., Hagao, K. and Kurosaki, K. 'Photographic Bibders., (Ed. H. Irie. et al) Japan, 1976, p 9
105. Ramanchandran, G.N. and Chandrasekharan, R. Biopolymers 1968, 6, 1649
106. Susi, H., Ard, J.S. and Carroll, R.J. Biopolymers 1971. 10, 1597
107. Luscher, M., Giovanoli, R., Hirter, P. Chimia 1973, 27, 112
108. Luescher, M., Ruegg, M. and Schindler, P. Biopolymers 1974, 13, 2489
109. Cerngross, O., Hermann, K. and Abitz, W. Biochem. Z. 1930,228, 409
110. Heidemann, E. and Keller, H. 1970, 65, 512
111. Chirgadze, Yu. N., Ven'yaminov, S. Yu. and Zimont, S.L. 'The State and Role of Water in Biological Objects', Nauka, 1967, p71
112. Millionova, M. 1., Esipova, N. C. and Rogulenkova, V. N. Biofizika 1972, 17, 575
113. Yannas, 1. V. and Tobolsky, A. V. Nature 1967, 215, 509
114. Godovskii, Yu. K., Mal'tseva, I.I. and Slonimskii, C.L. Vysokomol. Soedin. 1971, A13, 2768
115. Burdygina, G.I., Fridmann, I.M. and Kozlov, P. V. 'Photographic Gelatin II, (Ed. R.J. Cox), Academic Press, London and New York, 1976, p 179
116. Burdygina, G.I., Fridman, I.M., Kozlov, P. V. and Kargin, V. A. Vysokomol. Soedin. 1969, A11, 118
117. Fel'dman, R.I. Kolloid. Zh. 1959, 21, 499
118. Yannas,. I.V. and Tobolsky, A. V. Eur. Polym. J. 1968, 4, 257
119. Burdygina, G.I., Pron'kina, E. V., Radugina, Yu. E., Opel'boim, V. V. and Kozlov, P.V. Vysokomol. Soedin. 1976, A18, 1405
120. Burdygina, G.I., Kozlov, P.V. and Fridman, I.M. Proceedings of NIKFI No. 58, 23, 1970
121. Nakamura, K. Kolloid Z.,-Z. Polym. 1963, 189, 130
122. Burdygina, G.I., Fridman, I.M., Kozlov, P.V. and Kargin, V.A. Vysokomol. Soedin. 1969, All, 912
123. Burdygina, v Undzenas, A. I., Fridman, 1. M., Kozlov, P: V. and Kargin, V. A. Dokl. Akad. Nauk SSSR 1968, 178, 1360
124. Burdygina, G.I., Kozlov, P. V. and Kargin, V. A. Vysokomol. Soedin. 1972, A14, 383
125. Macsuga, D.D: Biopolymers 1972, II, 2521
126. Gillham, J. K. Appl. Polym. Symp. 1966, N 2, 45
127. Zaides, A. L. Kolloid. Zh. 1950, 12, 414
128. Dolgoplosk, B.A., Erusalimskii, B.L., Milovskaya, E.B. and Belonovskaya, G.P. Dokl. Akad. Nauk SSSR 1958, 120, 783
129. Fridman, I.M., Zenkina, L.I., Burdygina, G.I., and Kozlov, P.V. Adv. Sci. Photography 1977, 18, 120
130. Burdygina, G.I., Fauna, I. V., Fridman, I. M. and Kozlov, P. V. Vysokomol. Soedin. 1979, A21. 667
131. Burdygina, G.I., Falina, I.V., Fridman, I.M. and Kozlov, P.V. Vysokomol. Soedin. 1979, A21, 1188
132. Burdygina, G.I., Falina, I.V. and Kozlov, P.V. Vysokomol. Soedin. in press
133. Ostrikov, M. S., Dibrov, G.D. and Danilova, E. P. Dokl. Akad. Nauk SSSR 1958, 118, 751
134. Ostrikov, M. S., Rostovtseva, I.V., Dibrov, G.I. and Danilova, E.P. Kolloid. Z. 1960, 22, 443
135. Kargin, V. A. 'Current Problems of Polymer Science', MV. Lomonosov Moscow State University, 1962
136. Anokhin, V.V. Proceedings of Kiev Technological Institute of Legkaya Industriya, No. 10, 39, 1958
137. Grigoreva, N. V., Pchelin, V.A. and Rebinder, P.A. Dokl. Akad. Nauk SSSR 1961, 137, 889
138. Bergmann, M. and Jacobi, B. Kolloid Z. 1929, 49, 46
139. Burdygina, G.I., and Kozlov, P.V. Adv. Sci. Photography 1977, 18, 112
140. Burdygina, G.I., Aleksina, O.A., Fridman, I.M. and Kozlov, P.V. Proceedings of NIKFI, No. 58, 45, 1970
141. Burdygina, G.I., Fridman, I.M., Kozlov, P.V. and Kargin, V.A. Vysokomol. Soedin. 1969, A11, 632
142. Burdygina, G.I., Moteneva, Zh. F., Krasnova, N.P. and Kozlov, P.V. Vysokomol. Soedin. 1977, A19, 1158
143. Burdygina, G.I., Fauna, I.V. and Kozlov, P.V. Vysokomol. Soedin. 1979, A21, 1181
144. Burdygina, G.I., Moteneva, Zh. F., Boiko, O.K. and Kozlov, P. V. Kolloid. Z. 1975, 37, 9
145. Boiko, O.K., Burdygina, G.I., Ruzhitskaya, E. V. and Kozlov, P. V. Vysokomol. Soedin. 1979, B21, 382
146. Evseeva, V. K., Brainin, L.B., Perevezentseva, S.P., Biktimirov, R.S. and Zimkin, E.A. Z. Nauch. priklad. Fotograf. Kinemat. 1977, 22, 119
147. Evreinova, T.N. 'Concentration of Substances and Enzyme Action in Coacervates' Nauka, Moscow, 1966
148. Bello, J., Riese, H.C.A. and Vinograd, J.R. J. Phys. Chem. 1950. 60, 1299
149. Belle, J. and Vinograd, J.R. Nature 1958, 181, 273
150. Bello, J., Bello, H.R. and Vinograd, J.R. Biochim. Biophys. Acta 1962, 57, 222
151. Bello, J. Biochim. Biophys. Acta 1965, 109, 250
152. Kozlov, P.V. 'Polymers in Cinematography and Photography', Iskusstvo, 1960, p 60
153. Zimkin, E.A., Bobikova, T.M. and Klyuchevich, V.F. Z. Nauch. Priklad. Fotograf. Kinemat. 1975, 20, 233
154. Japanese patent No. 34515, 1975
155. Tourtellotte, B.D. and Williams, H.E. 'Recent Advances in Gelatin and Glue Research', 1958, p 246
156. Taylor, D.J. and Kragh, A.M. 'Photographic Gelatin II', (Ed. R. 3. Cox), Academic Press, London and New York, 1976, p 143
157. Burdygina, G.I., and Kozlov, P. V. Z. Nauch. Prik!. Fotograf. Kinemat 1977, 22, 35
158. Khismatullina, L.A., Levi, S. M. and Kukhtin, V.A. Vysokomol. Soedin. 1964, 6,473
159. Gaylord, N.G., Tomono, T. and Mandal, B.M. Polym. Lett. 1975, 13, 693
160. Denisova, A.A. and Sinyakova, L.I. Vysokomol. Soedin. 1968. A10, 357
161. Moshkina, T.M., Zimkin, E.A., Karnitskaya, R.I. and Klyuchevich, V.F. Uspekhi nauch. Fotograf. 1977. 18, 145
162. Kudaba, I.A., Chizhyunaite, E. Yu. and Yakubenaite, V.R. Vysokomol. Soedin. 1970, B12, 818
163. Kozlov, P.V., Yarysheva, L.M. and Bakeev, N.F. Trudy NIKFI 1974, 73, 59
164. Yarysheva, L.M., Averbukh, M.Z., Bakeev, N.F. and Kozlov, P.V. Vysokomol. Soedin. 1974, A16, 1807